硫代基夫碱叠氮基糖原料中间体的合成
2013-10-09赵莲冯霞李锐陈华
赵莲,冯霞,李锐,陈华
(1.河北大学化学与环境科学学院河北省化学生物学重点实验室,河北保定 071002;2.河北大学人事处,河北保定 071002;3.重庆第二师范学院生物与化学工程系,重庆 400067)
基夫碱(kifunensine,图1)是从放线菌(KitasatosporiakifunensineNo.9482)中分离提取得到的天然生物碱,是一种并有双羰基咪唑啉结构的多羟基哌啶类双环氮杂糖[1].基夫碱可以选择性的抑制I类甘露糖苷酶,抑制活性IC50值达到0.05μmol/L,而对Ⅱ类甘露糖苷酶没有抑制作用[2].由于甘露糖苷酶在病毒感染、糖尿病和肿瘤转移等重大疾病的发生、发展过程中扮演有重要角色[3],通过对基夫碱的结构改造,获得高效糖苷酶抑制剂,是研发新型抗肿瘤、抗病毒及抗糖尿病的糖类药物的有效途径之一[4].基于生物电子等排原理,实验室以甘露糖为起始原料,利用Staudinger/aza-Wittig/cyclization串联反应及2次Pummerer重排反应合成了硫原子取代氮原子的硫代基夫碱(thiokifunensine,式1),化合物对HIV逆转录酶有显著抑制活性[5].
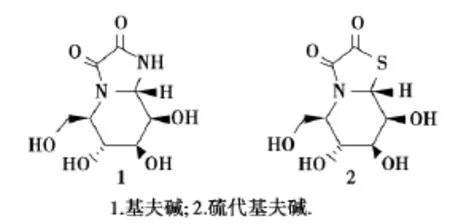
图1 基夫碱和硫代基夫碱结构Fig.1 Structures of kifunensine and thiokifunensine
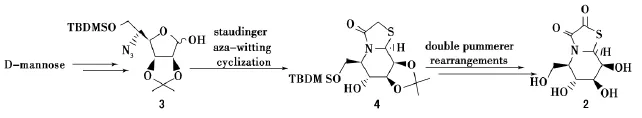
式1 硫代基夫碱的合成Scheme 1 Synthesis of thiokifunensine
硫代基夫碱合成的关键是叠氮基糖原料3的制备.以甘露糖为起始原料,其间涉及到5位羟基的翻转.文献[6]报道,利用Mitsunobu反应可以实现5位羟基的翻转(式2),但随后需要在碱性条件下脱除反应带入的三氟乙酰基,这对1位碱敏感的苯甲酰基会产生较大影响.选择性脱除反应不易控制,且产率低.因此,笔者尝试利用条件温和的亚硝酸盐介导的Lattrell-Dax羟基翻转反应[7],探讨叠氮基糖原料3的合成(式3),为硫代基夫碱的合成提供一条新的路线.
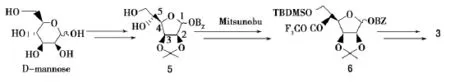
式2 利用Mitsunobu反应实现5-羟基的翻转Scheme 2 5-Hydroxy inversion by Mitsunobu reaction
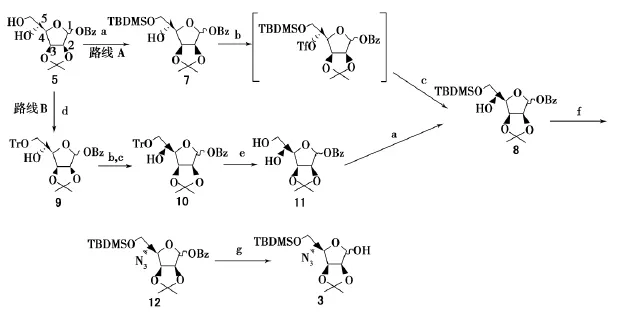
式3 利用亚硝酸盐介导的Lattrell-Dax反应合成化合物3 Scheme 3 Synthesis of 3by nitrite-mediated Lattrell-Dax reaction
1 实验部分
1.1 材料
熔点由SGWRX-4显微熔点仪(温度计未校正)测定;旋光由SGWRX-1自动旋光仪测定;核磁共振谱用BRUKER AC-P600(600MHz)型核磁共振仪测定,TMS为内标;高分辨质谱(ESI)用FTICR-MS(Ionspec 7.0T)型质谱仪测定.层析用硅胶(40~45μm)为青岛海洋化工厂产品.本文所用其他试剂均为分析纯,无水试剂均按常规方法处理,水为二次蒸馏水.
1.2 以化合物5为原料按照路线A合成8
1.2.1 1-O-苯甲酰基-2,3-O-异丙基-6-O-叔丁基二甲基硅基呋喃D甘露糖(7)的合成
将1-O-苯甲酰基-2,3-O-异丙基呋喃D甘露糖化合物5[8](9.7g,0.03mol)加入100mL圆底烧瓶中,抽真空,N2保护,加入无水吡啶100mL将其溶解,将溶液温度降至0℃,搅拌下加入叔丁基二甲基氯硅烷(TBDMSCl,4.95g,0.03mol).薄层色谱(TLC)监测反应,8h后,反应完毕.加入300mL乙酸乙酯混匀,用1mol/L稀盐酸洗至略显酸性,饱和食盐水200mL×4洗至中性,加入无水硫酸镁干燥过夜,抽滤除去无水硫酸镁,减压蒸除去乙酸乙酯,柱层析(V(石油醚)∶V(乙酸乙酯)=3∶1)得12.1g化合物7白色固体,产率92%.mp.83~84℃;+0.004(c 1.0,CHCl3);1H NMR(600MHz,CDCl3)δH:7.99(d,J=7.2Hz,2H),7.58(t,J=7.2Hz,1H),7.44(t,J=7.8Hz,2H),6.39(s,1H),5.01(dd,J=5.4,3.6Hz,1H),4.87(d,J=6.0Hz,1H),4.15(dd,J=9.0,3.6Hz,1H),3.99~4.03(m,1H),3.85(dd,J=10.2,3.6Hz,1H),3.76(dd,J=10.2,4.2Hz,1H),2.75(d,J=6.6Hz,1H),1.52(s,3H),1.38(s,3H),0.81(s,9H),0.03(s,3H),-0.01(s,3H);13C NMR(150MHz,CDCl3)δC:164.8,133.4,129.7,128.4,113.2,101.4,85.0,81.0,79.8,69.0,64.0,26.1,25.7,24.8,18.2,-5.4,-5.6;HRESIMS:calcd for C22H34O7SiNa([M+Na]+)461.1951,found:461.1969.
1.2.2 1-O-苯甲酰基-2,3-O-异丙基-6-O-叔丁基二甲基硅基呋喃D古洛糖(8)的合成
将化合物7(15.3g,0.04mol)加入500mL圆底烧瓶中,加入200mL二氯甲烷溶解,降至-40℃搅拌下加入8mL无水吡啶,滴加三氟甲磺酸酐(Tf2O,11.7mL,0.04mol),氮气的保护下搅拌,TLC监测反应,1h后,反应完全.加入200mL二氯甲烷混匀,用1mol/L稀盐酸洗至略显酸性,饱和食盐水250mL×4洗至中性,加入无水硫酸镁干燥,抽滤除去无水硫酸镁,减压蒸除去二氯甲烷,得橙色黏稠液体.用200mLN,N-二甲基酰胺(DMF)将其溶解,降至0℃搅拌下分批加入NaNO2(12.3g,0.18mol),加完撤去冰浴,氮气保护下搅拌反应,TLC监测反应,4h后,反应完毕.加入1mol/L稀食盐水50mL,用乙酸乙酯100mL×3萃取,混合乙酸乙酯部分,用200mL×5稀食盐水洗,饱和食盐水200mL×2洗,加入无水硫酸镁干燥过夜,抽滤除去无水硫酸镁,减压蒸除乙酸乙酯,柱层析V(石油醚)∶V(乙酸乙酯)=3∶1)得7与8的混合物8.6g,两步产率56%,异构体比例为1∶1.
1.3 以化合物5为原料按照路线B合成8
1.3.1 1-O-苯甲酰基-2,3-O-异丙基-6-O-三苯甲基呋喃D甘露糖(9)的合成
将化合物5(4.7g,0.01mol)加入250mL圆底烧瓶中,用50mL二氯甲烷溶解,并加入1.6mL无水吡啶作为缚酸剂,0.1g 4-二甲氨基吡啶(DMAP)为催化剂,在冰水浴下,搅拌下加入三苯基氯甲烷(TrCl,5.1g,0.01mol)的二氯甲烷溶液,加完后升至室温,氮气保护下搅拌.TLC监测反应,4h后,反应完全.加入200mL二氯甲烷混匀,用1mol/L稀盐酸洗至略显酸性,饱和食盐水150mL×3洗至中性,加入无水硫酸镁干燥过夜,抽滤,减压蒸除二氯甲烷.残留物用甲醇溶解,析出6.2g化合物9白色固体,产率76%.mp.164~166℃;+0.014(c 1.0,CHCl3);1H NMR(600MHz,CDCl3)δH:8.02(d,J=4.8Hz,2H),7.58(t,J=7.2Hz,1H),7.22~7.45(m,17H),6.43(s,1H),5.03(dd,J=6.0,3.6Hz,1H),4.88(d,J=6.0Hz,1H),4.36(dd,J=7.8,3.6Hz,1H),4.14~4.17(m,1H),3.42~3.43(m,2H),2.83(d,J=6.0Hz,1H),1.53(s,3H),1.40(s,3H);13C NMR(150MHz,CDCl3)δC:164.9,143.7,133.4,129.7,128.6,128.4,127.8,127.0,113.3,101.4,86.7,85.0,81.4,80.0,77.2,77.0,76.8,68.9,64.7,26.0,24.8;HRESIMS:calcd for C35H34O7Na([M+Na]+)589.2182,found:589.2161.1.3.2 1-O-苯甲酰基-2,3-O-异丙基-6-O-三苯甲基呋喃D古洛糖(10)的合成
将化合物9(20g,0.04mol)加入500mL圆底烧瓶中,加入200mL二氯甲烷溶解,降至-40℃搅拌下加入8mL无水吡啶,滴加Tf2O(11.7mL,0.04mol),氮气的保护下搅拌.升温至-25℃继续搅拌.TLC监测反应,1h后,反应完全.加入200mL二氯甲烷混匀,用1mol/L稀盐酸洗至略显酸性,饱和食盐水250mL×4洗至中性,加入无水硫酸镁干燥,抽滤除去无水硫酸镁,减压蒸除二氯甲烷,得橙色黏稠液体.用200mL DMF将其溶解,降至0℃搅拌下分批加入NaNO2(12.3g,0.18mol),加完撤去冰浴,氮气保护下搅拌反应.TLC监测反应,4h后,反应完全.加入1mol/L稀食盐水50mL,加入100mL×3乙酸乙酯萃取,混合乙酸乙酯部分,用200mL×5稀食盐水洗,饱和食盐水200mL×2洗,加入无水硫酸镁干燥过夜,抽滤除去无水硫酸镁,减压蒸除去乙酸乙酯,柱层析(V(石油醚)∶V(乙酸乙酯)=5∶1)得10.4g化合物10白色固体,两步产率56%.mp.111~114℃;+0.041(c 1.0,CHCl3);1H NMR(600MHz,CDCl3)δH:8.01(d,J=7.2Hz,2H),7.59(t,J=7.8Hz,1H),7.23~7.48(m,17H),6.46(s,1H),4.86(d,J=6.0Hz,1H),4.74(dd,J=5.4,3.0Hz,1H),4.45(dd,J=6.0,3.6Hz,1H),4.26(d,J=5.4 Hz,1H),3.47(dd,J=9.6,5.4Hz,1H),3.31(dd,J=9.6,4.8Hz,1H),2.90(s,1H),1.50(s,3H),1.30(s,3H);13C NMR(150MHz,CDCl3)δC:164.9,143.8,133.4,129.7,128.6,128.4,127.8,127.0,113.2,101.2,86.7,85.6,82.2,80.0,69.7,63.9,26.0,24.5;HRESIMS:calcd for C35H34O7Na([M+Na]+)589.2182,found:589.2167.
1.3.3 化合物8的合成
将化合物10(300mg,0.53mmol)加入50mL圆底烧瓶中,加入10mL 70%(体积分数)AcOH溶解,油浴50℃搅拌.TLC监测反应,16h后,反应完全.加入Na2CO3将溶液中和至中性,乙酸乙酯10mL×3萃取.合并有机相,饱和食盐水20mL×3洗涤,加入无水硫酸镁干燥过夜,抽滤除去无水硫酸镁,减压蒸除酸乙酯,柱层析(V(石油醚)∶V(乙酸乙酯)=1∶1)得128mg白色固体(化合物11,1-O-苯甲酰基-2,3-O-异丙基呋喃D古洛糖),产率75%.取化合物11(8g,0.02mol)加入250mL圆底烧瓶中,加入无水吡啶100mL溶解,将溶液温度降至0℃,搅拌下加入TBDMSCl(4.7g,0.03mol),TLC监测反应,8h后,反应完全.加入300mL乙酸乙酯混匀,用1mol/L稀盐酸洗至略显酸性,饱和食盐水200mL×4洗至中性,加入无水硫酸镁干燥过夜,抽滤除去无水硫酸镁,减压蒸除去乙酸乙酯,柱层析(V(石油醚)∶V(乙酸乙酯)=3∶1)得10.0g化合物8白色固体,产率92%.mp.75~76℃;[α]28D+0.047(c 1.0,CHCl3);1H NMR(600MHz,CDCl3)δH:8.00(d,J=7.2Hz,2H),7.57(t,J=7.2Hz,1H),7.43(t,J=7.8Hz,2H),6.45(s,1H),4.87~4.89(m,2H),4.30(dd,J=5.4,3.0Hz,1H),4.07~4.12(m,1H),3.75~3.81(m,2H),2.94(d,J=3.0Hz,1H),1.52(s,3H),1.34(s,1H),0.86(s,9H),0.06(s,3H),0.03(s,3H);13C NMR(150MHz,CDCl3)δC:164.9,133.3,129.7,128.3,113.3,101.1,85.7,81.4,80.2,70.7,63.5,26.0,25.8,24.6,18.2,-5.4;HRESIMS:calcd for C22H34O7SiNa([M+Na]+)461.195 1,found:461.196 3.
1.4 1-O-苯甲酰基-2,3-O-异丙基-5-叠氮基-6-O-叔丁基二甲基硅基呋喃D甘露糖(12)的合成
将化合物8(2.4g,5.46mmol)加入100mL圆底烧瓶中,加入20mL二氯甲烷溶解,降至-40℃搅拌下加入1.32mL无水吡啶,滴加Tf2O(1.90mL,6.0mmol),氮气的保护下搅拌.TLC监测反应,1h后,反应完全.加入100mL二氯甲烷混匀,用1mol/L稀盐酸洗至略显酸性,饱和食盐水洗50mL×3至中性,加入无水硫酸镁干燥,抽滤除去无水硫酸镁,减压蒸除去二氯甲烷,得橙色黏稠液体.用50mL DMF将其溶解,降至0℃搅拌下加入NaN3(810mg,10.9mmol),加完撤去冰浴,氮气保护下搅拌反应.TLC监测反应,8h后,反应完全.加入1mol/L稀食盐水20mL,加入50mL×4乙酸乙酯萃取,混合乙酸乙酯部分,用30mL×5稀食盐水洗,饱和食盐水30mL×2洗,加入无水硫酸镁干燥过夜,抽滤除去无水硫酸镁,减压蒸除去乙酸乙酯,柱层析(V(石油醚)∶V(乙酸乙酯)=10∶1)得1.7g化合物12黄色油状液体,2步产率68%+0.052(c 1.0,CHCl3);1H NMR(600MHz,CDCl3)δH:8.00(d,J=8.4Hz,2H),7.58(t,J=7.8Hz,1H),7.44(t,J=7.8Hz,2H),6.38(s,1H),4.95(dd,J=5.4,3.6Hz,1H),4.87(d,J=5.4Hz,1H),4.19(dd,J=10.2,3.6Hz,1H),4.03(dd,J=10.8,3.2Hz,1H),3.84(dd,J=11.4,6.0Hz,1H),3.68~3.71(m,1H),1.52(s,3H),1.39(s,3H),0.81(s,9H),0.04(s,3H),0.00(s,3H);13C NMR(150MHz,CDCl3)δC:164.7,133.4,129.7,128.4,113.3,101.2,84.8,79.5,79.4,63.5,60.4,26.1,25.6,24.9,18.1,-5.5,-5.7;HRESIMS:calcd for C22H33N3O6SiNa([M+Na]+)486.2016,found:486.200 9.
1.5 2,3-O-异丙基-5-叠氮基-6-O-叔丁基二甲基硅基呋喃D甘露糖(3)的合成
将化合物12(1.5g,3.2mmol)加入100mL圆底烧瓶中,加入45mL无水甲醇溶解,室温搅拌下加入CH3ONa(87.5mg,3.2mmol),氮气保护搅拌,TLC监测反应,2h后,反应完全.加入阳离子交换树脂将反应液中和至中性,抽滤除去阳离子交换树脂,加入硅胶蒸干,柱层析(V(石油醚)∶V(乙酸乙酯)=10∶1),得无色透明液体816mg化合物3,产率71%.与标准品对照,多种展开剂展开,Rf值不变,波谱数据与文献报道一致.
2 结果与讨论
Lattrell-Dax反应主要用于糖羟基构型的翻转,是SN2取代反应.该反应首先是羟基的三氟甲磺酰(Tf)化,随后被亚硝酸根离子)取代水解,将羟基构型翻转.反应条件温和,往往得到单一构型的异构体,但受相邻羟基构型及其保护基的影响较大[9].笔者首先尝试了化合物7(硅基TBDMS保护6位羟基)的Lattrell-Dax反应(路线A,式3).5位羟基上的三氟甲磺酰基化在-40℃下可反应完全.薄层色谱(TLC)监测化合物7已全部转换为其Tf化产物.该产物不稳定,在室温下放置,易发生消除反应.因此,此反应在后处理时要快速,并控制温度在20℃以下.处理完成后,直接投到下一步亚硝酸钠(NaNO2)主导的羟基构型翻转反应中.但反应完成后,得到了化合物7(最初原料)和8(翻转产物)的异构体混合物,比例为1∶1且无法分离.
考虑到可能是6位硅基保护基的问题,笔者进一步尝试了化合物9(三苯甲基Tr保护6位羟基)的Lattrell-Dax反应(路线B,式3).以化合物5为原料,首先对其6位羟基进行Tr保护.按常规方法,以无水吡啶为溶剂,室温搅拌36h,但反应完成后除了生成化合物9,还生成了大量的三苯甲醇.由于三苯甲醇的溶解性与化合物9类似,无法使用重结晶的方法提纯化合物9.而采用柱层析,三苯甲醇与产物在淋洗剂石油醚-乙酸乙酯体系中溶解性不好,导致柱层析过程缓慢且化合物在柱子上易析出,使硅胶柱堵塞.因此,笔者对反应条件进行了改进,使用无水二氯甲烷溶解原料,加入少量无水吡啶(Pry)作为缚酸剂,并加入4-二甲氨基吡啶(DMAP)催化反应.在冰水浴下,搅拌下缓慢滴加TrCl的二氯甲烷溶液,滴加完毕撤掉冰浴,搅拌反应4h即反应完全.反应生成的三苯甲醇量极少,反应液中和、萃取浓缩后,加入甲醇即可析出纯净的化合物9白色固体.
随后,对化合物9进行三氟甲磺酰(Tf)化,在-40℃下原料反应不完全,需升温至-25℃才会反应完全.可能是三苯甲基(Tr)较叔丁基二甲基硅基(TBDMS)空间位阻大,导致反应活性降低.进一步使用NaNO2将5位羟基构型成功翻转,得到单一产物化合物10.反应过程中,加入NaNO2后大量放热,即使在冰浴条件下也会使原料(Tf化原料不稳定)发生消除,影响产率(30%).为了提高产率,将NaNO2分批加入反应液中,冰浴中剧烈搅拌,产率最终提高到52%.由于Tr基团在后续的实验(Staudinger/aza-Wittig/cyclization串联反应)中易脱除,因此将Tr基团脱保护后(化合物11),再使用TBDMS保护6位伯羟基得到化合物8.对三苯甲基的脱除条件进行了探讨(表1),最终确定70%(体积分数)CH3COOH和50℃的反应条件(编号3),增加酸度或提高反应温度,产率均会降低.

表1 三苯甲基的脱除条件探讨Tab.1 Studies of Tr deprotection
最后,化合物8经叠氮化、脱除1位苯甲酰基(Bz),以17.6%的总收率(从原料5开始计)合成了目标产物3.较文献报道方法(Mitsunobu反应)[6],Lattrell-Dax反应在羟基翻转中更易于控制,且收率高.同时合成过程中,Lattrell-Dax反应条件的有益探讨,将有助于该反应在糖差向异构体合成中的进一步应用.
[1] KAYAKIRI H,TAKESE S,SHIBATA T,et al.Structure of kifunensine,a new immunomodulator isolated from an actinomycete[J].J Org Chem,1989,54:4015-4016.
[2] ELBEIN A D,TROPEA J E,MITCHELL M,et al.Kifunensine,apotent inhibitor of the glycoprotein processing mannosidase I[J].J Biol Chem,1990,265:15599-15605.
[3] KARAVEG K,MOREMEN K W.Energetics of substrate binding and catalysis by class I(glycosylhydrolase family 47)α-mannosidases involved inN-glycan processing and endoplasmic reticulum quality control[J].J Biol Chem,2005,280:29837-29848.
[4] HERING K W,KARAVEG K,MOREMEN K W,et al.A practical synthesis of kifunensine analogues as inhibitors of endoplasmic reticulumα-mannosidase I[J].J Org Chem,2005,70:9892-9904.
[5] CHEN Hua,LI Rui,LIU Zhenying,et al.Synthesis of kifunensine thioanalogs and their inhibitory activities against HIV-RT andα-mannosidase[J].Carbohydra Res,2013,365:1-8.
[6] COSME G F,RAIMUNDO F,CONCEPCIóN C G,et al.Fragmentation of carbohydrate anomeric alkoxy radicals.Synthesis of polyhydroxy piperidines and pyrrolidines related to carbohydrates[J].J Org Chem,2001,66:1861-1866.
[7] WANG Guangfa,ZHANG Wei,LU Zhichao,et al.Convenient synthesis of anN-glycan octasaccharide of the bisecting type[J].J Org Chem,2009,74:2508-2515.
[8] SUHARA Y,KITTAKA A,KURIHARA M,et al.Design and efficient synthesis of new stable 1α,25-dihydroxy-19-norvitamin D3analogues containing amide bond[J].Bioorg Med Chem Lett,2002,12:3533-3536.
[9] DONG Hai,PEI Zhichao,RAMASTRÖM O.Stereospecific ester activation in nitrite-mediated carbohydrate epimerization[J].J Org Chem,2006,71:3306-3309.
