腓骨肌萎缩症(CMT)1型的临床、神经电生理及CMT1A型基因诊断方法的研究
2013-09-20曹秉振
史 磊, 曹秉振
腓骨肌萎缩症(charcot-marie-tooth disease,CMT)又称遗传性运动感觉神经病(hereditary motor and sensory neuropathy,HMSN),是最常见的遗传性周围神经疾病,发病率为1/2500[1]。其主要特征为慢性进行性加重的肢体远端无力、肌萎缩和感觉损害。CMT的遗传方式主要为常染色体显性遗传(AD),也可见常染色体隐性遗传(AR)及X连锁显性或隐性遗传(XD或 XR)。根据电生理和病理特征,CMT可分为CMT1(脱髓鞘型)、CMT2(轴索型)和其他较罕见的类型[2]。目前,分子生物学研究已经发现了近40个不同的CMT致病基因位点,20多个致病基因已被克隆。临床上最常见的类型为CMT1型,其中50% ~70%的CMT 1患者及90%的散发病例为 CMT1A型[3],多由17号染色体短臂11.2区(17p11.2)包含周围髓鞘蛋白 PMP 22基因在内的1.5MB的正向串联重复突变所致。
我们收集了5例临床基本确诊为CMT1型的患者,分析比较他们的临床和电生理特点,并分别应用等位基因特异性PCR和多重连接探针扩增技术(MLPA)两种方法对5例患者进行PMP22基因重复突变检测,初步验证两种方法所得结果的一致性,指导临床开展相关基因筛查和诊断的基本程序以进一步确定CMT1A型的诊断。
1 对象与方法
1.1 研究对象 5例CMT1型患者均来源于济南军区总医院神经内科门诊及住院的病例。所有患者均由神经内科专科医生进行详细的病史询问和体格检查,符合Harding于1980年制定的诊断标准[4]。所有受试者均征得本人知情同意。
1.2 神经电生理检查 主要为运动和感觉神经传导速度的检查,上肢包括正中神经和尺神经,下肢运动神经主要检查腓总神经和胫神经,感觉神经为腓浅神经和腓肠神经。
1.3 基因突变检测
1.3.1 等位基因特异性PCR检测 抽取患者外周静脉血3~5ml,用EDTA抗凝,Promega试剂盒提取全血标本基因组DNA。在PMP22基因重复片段的远端和近端的区域内,设计出等位基因特异性引物,使其可扩增出患者特异性重复连接片段[5],PCR反应体系为:总体积50μl,其中 dNTP Mixture(2mmol/L)5μl,10 × PCR buffer 5μl,TaqDNA 聚合酶0.25U,基因组 DNA2μl,上下游引物(5μmol/L)各5μl,三蒸水 27.75μl。反应条件为在 GeneAmp PCR system 2400热循环仪上94℃预变性5min,然后 94℃变性 30s,56℃退火 1min,72℃延伸 3min,25次循环,最后72℃延伸5min,4℃保存。反应完成后取5μl PCR产物与1μl溴乙锭混合,0.8%琼脂糖凝胶电泳(加核酸染色液)检测产物。
1.3.2 MLPA检测 MLPA检测试剂盒 SALSA P033 购自 MRC-Holland,取基因组 DNA 1μl,加TE稀释至5μl,在热循环仪上98℃变性5min后冷却至 25℃,分别加入 1.5μl P033探针混合物及1.5μl MLPA buffer,小心混匀后 95℃ 变性 1min,60℃下杂交16h~20h。杂交后在54℃下加入连接混合物(25μl三蒸水,3μl连接缓冲液 A,3μl连接缓冲液 B,1μl连接酶)孵育15min,然后98℃加热5min以灭活连接酶,待温度降至20℃后,加入PCR反应混合物(7.5μl三蒸水,2μlSALSA PCR 引物混合物,0.5μl SALSA 聚合酶),PCR 反应条件为:95℃变性30s,60℃ 退火 30s,72℃ 延伸 60s,循环 35 次,72℃延伸 20min,取0.7μl PCR 反应产物加入 0.2μl LIZ-500标记物及9μl甲酰胺(LIZ-500及甲酰胺均为美国ABI公司生产),80℃变性2min后立即置冰架上冷却,最后用毛细管凝胶电泳检测(在ABI3130基因测序仪上进行),程序结束后导出数据,结果用Coffalyser软件分析。
2 结果
2.1 临床特征 4例患者20岁前发病,其中2例有家族史,1例患者30岁左右发病,无家族史。所有患者均表现进行性四肢远端无力伴肌萎缩,下肢重于上肢,4例患者四肢远端感觉减退,1例患者感觉减退不明显。所有患者均出现腱反射减弱或消失,2例患者出现足弓变形。
2.2 神经电生理检查 2例患者上肢正中神经传导速度严重降低(<25m/s),3例患者上肢正中神经传导速度明显降低(<38m/s),4例患者下肢腓总神经传导速度严重降低(<20m/s),1例患者明显降低(<38m/s)。4例患者所检上下肢感觉神经未引出动作电位,1例患者所检感觉神经传导速度明显降低。患者所检神经波幅均有不同程度降低。
2.3 基因突变分析
2.3.1 等位基因特异性PCR产物检测结果1例患者出现了经特异引物扩增的特异性短片段重复的异常条带,其余4例患者未出现异常条带。
2.3.2 MLPA检测分析 运行软件Coffalyser分析得出:4例患者的PMP22基因全部在正常范围内,只有1例患者PMP22基因的几乎所有外显子均在正常范围上限以上,表明这例患者存在PMP22基因的重复突变(图1为PMP22基因重复突变患者的分析结果,正常范围结果图略)。
3 讨论
CMT1型的患者发病年龄较早,常儿童期起病,患者表现比同龄儿童跑跳困难,而后出现进行性四肢无力和肌萎缩,严重时可见爪型手和弓形足。此次收集的病例临床症状相对单一,未见有其他报道中提到的神经功能重度受损、,如伴锥体束征、伴神经性耳聋等相关症状出现[6],说明CMT1临床表型的变异较大。
临床上常根据电生理和病理特点大致分为CMT1型(脱髓鞘型)和CMT2型(轴索型),电生理主要依据神经传导速度(NCV)减慢的程度和波幅是否降低未区分,脱髓鞘型一般表现为正中神经NCV<38m/s且波幅无明显降低,而轴索型出现波幅明显降低但正中神经NCV >38m/s。本研究中患者均出现运动神经传导速度减慢,其中2例较严重病例正中神经NCV<25m/s,下肢腓总神经NCV<20m/s,且出现继发性波幅降低。患者虽表现四肢末端感觉障碍较轻但是感觉神经的动作电位较难引出。可见神经传导速度降低往往会继发波幅降低,同样波幅降低也会继发神经传导速度减慢,所以很多情况根据电生理不好明确区分。神经病理活检可以明确患者为何种类型,但损伤较大,患者接受程度较低。相比之下基因检测的准确性高,基本无损伤,在病程早期即可做出明确诊断,有较高的临床应用价值。
本研究中采用了两种方法检测患者的PMP22基因是否存在短片段重复突变,等位基因特异性PCR是直接对PMP22基因交换热点区设计引物扩增出特异性连接片段,Kon-Ping Lin等根据已被共识的近端和远端CMT1A-REP序列设计出了PMP22等位基因特异性引物[7],其优点是简便易行,对操作条件及成本要求较低,但其灵敏度和准确性仍需进一步验证。而新技术MLPA-CMT1A试剂盒的探针基本覆盖了PMP22基因外显子的所有热点区域,且能同时检测基因的重复与缺失,敏感度及准确度较等位基因特异性PCR更高,且MLPA与荧光定量PCR、STR等方法相比也具有一定的优势:(1)可同时检测多基因多外显子的重复或缺失。(2)对DNA模板定量及纯度要求较低。当然,MLPA也有自身的缺点和限制,比如价格稍昂贵、对实验室的配置条件要求较高等。因此,两种方法对特异性短片段重复突变的检测都具有不可替代的作用和意义。建议在临床患者初筛或基础设施比较简单的实验室条件下,可以先采用等位基因特异性PCR进行检测,对可疑阳性患者再进一步选择灵敏度和准确度更高的MLPA确定诊断。
CMT涉及的致病基因较多,仅CMT1型相关的基因就有6个,基因检测虽然准确性高但要确定具体的致病基因可能需要多次实验,未检测出PMP22基因重复突变的患者很有可能存在其他基因的突变,需要进一步实验验证。本实验综合研究了CMT1型患者的临床和电生理特征,同时研究了CMT1A型基因突变的检测方法,旨在指导临床根据电生理特征及PMP22基因重复突变的检测更好地确定CMT1A型的诊断。
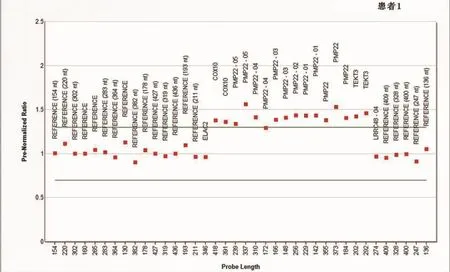
图1 PMP22基因重复突变患者的分析结果
[1]Berger P,Young P,Suter U,etal.Molecular cell biology of Charcot-Marie-Tooth disease[J].Neurogenetics,2002,4(1):1-15.
[2]Street VA,Bennett CL,Goldy JD.Mutation of a putative protein degradation gene LITAG/SIMPLE in Charcot-Marie-Tooth disease LC[J].Neurology,2003,60:22.
[3]萧剑锋,唐北沙,夏家辉,等.聚合酶链反应技术在腓骨肌萎缩症基因诊断中的应用[J].中华医学杂志,2001,81:138.
[4]Harding AE,Thomas PK.The clinical features of hereditary motor and sensory neutopathy types1 and 2[J].Brain,1980,103(2):258-280.
[5]Lin KP,Chou CH,Lee HY,etal.Allele-specific.All-or-None PCR product diagnostic strategy for Charcot-Marie-Tooth 1A and hereditary neuropathy with liability to pressure palsies[J].JChin Med Assoc,2006,69(2):68-73.
[6]郭 鹏,宋福聪,王相斌,等.常染色体显性遗传腓骨肌萎缩症的临床与基因突变特点[J].中风与神经疾病杂志,2011,28(8):705-707.
[7]Zmurovic N,Millc V,Dackovic J,etal.Analysisi ofmutation in the chromosome 17p11.2 region in patients with Charcot-Marie-Tooth type 1 disease and in patients with tomaculous neuropathy[J].Srp Arh Celok Lek,2002,130(3-4):59-63
