沉淀-柱层析法纯化茶多酚及儿茶素含量测定
2013-09-05朱宇惠张继
朱宇惠,张继
(1.潞安职业技术学院,山西长治 046204;2.甘肃特色植物有效成分制品工程技术研究中心,甘肃兰州 730073)
茶已被人们誉为世界性饮料,茶叶的一些生理活性已被人们逐步开发利用。它的活性成分茶多酚(Tea Polyphenols,简称TP)又称茶鞣或茶单宁,是茶叶中多酚类物质的总称,主要包括儿茶素类、黄酮类、花色素类和酚酸类四种物质[1]。儿茶素主要指儿茶素(C)、表儿茶素(EC)、没食子儿茶素(GC)、表没食子儿茶素(EGC)、表儿茶素没食子酸脂(ECG)、没食子儿茶素没食子酸酯(GCG)和表没食子儿茶素没食子酸脂(EGCG)七种儿茶素[2]。
本实验利用沉淀法去除茶多酚粗品中咖啡碱等杂质,再用柱层析法精制茶多酚。最后采用高效液相和福林酚试剂比色法的联用对茶多酚产品进行了定性、定量分析。
1 材料与方法
1.1 材料及试剂
无水乙醇、浓盐酸、氯化锌、碳酸氢钠、石油醚、乙酸乙酯、甲酸均为分析纯;茶多酚粗品购自天水维康生物工程有限公司。
1.2 仪器设备
PHS-3C精密PH计:上海安亭吉路149号雷磁仪器厂;RE-2000A旋转蒸发仪:保定市阳光仪器厂;SHD-ⅢN型循环水式多用真空泵:保定高新区阳光科教仪器厂;DF-101S焦热式恒温加热磁力搅拌器:巩义市英峪高科仪器厂;柱层析硅胶(200目~300目);层析柱(口径1.5cm);紫外可见分光光度计:北京莱伯泰科仪器有限公司;ES5000-1电子天平:沈阳龙腾电子有限公司。
1.3 方法

金属沉淀法包括下述沉淀实验,酸转溶实验,乙酸乙酯萃取实验和减压蒸馏实验四部。
1.3.1 茶多酚粗品的预处理
为了使茶多酚最大范围的溶解,并为下一步的沉淀反应提供最佳的沉淀环境,该试验选用60%乙醇溶液溶解茶多酚粗品,并配成浓度为100 mg/mL样品溶液。磁力加热搅拌器调至50℃,转速为40 r/min,搅拌样品溶液[3]。30 min后停止搅拌并过滤,滤液即为茶多酚样品溶液。
1.3.2 沉淀实验
向茶多酚预处理溶液中加入复合沉淀剂(Zn2+、Al3+),控制反应的温度为45℃,并用1.0 mol/L NaHCO3调节反应pH在5.5~6.6范围内,随之产生大量黄色泥土状沉淀。静置10 min,当pH无明显变化时,取上层清夜2 mL~3 mL,加1滴NaHCO3,若无黄色浑浊现象则表明沉淀完全[4-6]。
1.3.2.1 沉淀剂的选择
常用沉淀剂:①重金属碱式盐,如Pb(OH)Ac、Cu(OH)Ac等,但 Pb,Cu,Ba毒性大,不宜使用;② 氢氧化物,如Ca(OH)2,这类沉淀剂属于强碱,会氧化茶多酚,而且Ca(OH)2为微溶物,沉淀效果也不好;③金属盐离子,如 Ca2+、Zn2+、Al3+、Mg2+、Ba2+和 Fe3+等。三类沉淀剂相比较之下,金属离子沉淀剂价廉,而且在一定的pH下能够较好地沉淀,且稀酸可转溶。沉淀剂的选择结果见表1。

表1 沉淀剂的选择Table 1 The selection of the precipitation
如表1所示,锌离子和铝离子对茶多酚的沉淀具有协同作用,茶多酚的沉淀率最高,而且在相对偏酸性的环境下沉淀,不易使茶多酚氧化。因此复合沉淀剂比单一沉淀剂的沉淀效果要好。
1.3.2.2 溶液pH的选择
复合沉淀剂的最佳沉淀pH是5.5~6.6,如果在更酸的环境下,沉淀物会变性,呈现黏稠的浆糊状,根本无法抽滤。因此为保证沉淀效果最佳,加沉淀剂前先将溶液pH调至5.5,然后将酸度计插入溶液中。不断少量加复合沉淀剂,根据酸度计的示数,加碱使酸度控制在5.5~6.6范围内。还有,为了提高反应效率,复合沉淀剂按质量比 m(AlCl3)∶m(ZnCl2)=1 ∶2 称量好混匀,用蒸馏水溶解成饱和溶液备用。
1.3.2.3 碱的选择
在上一步调节pH时,需要用碱来调节。NaOH溶液适宜浓度为0.1 mol/L~0.5 mol/L,浓度过低会使操作体积过大,浓度过高会因茶多酚的局部氧化严重而降低提取率。通过试验,本试验采用浓度为1.0 mol/L的NaHCO3溶液。
1.3.2.4 温度的选择
在加入沉淀剂的过程中,由于咖啡碱会与茶多酚产生共沉淀作用,而使茶多酚的提取率降低。相应地提高温度会增大咖啡碱的溶解度,使共沉淀作用减小,但是温度过高会导致茶多酚的氧化。通过试验,本试验采用45℃为最佳沉淀温度。
1.3.3 酸转溶实验
由于复合沉淀剂选择的是氯盐,为了减少溶液中离子的种类,在酸转溶时选择稀盐酸,而不选硫酸。酸浓度太小会需要大量的溶剂;浓度过大,酸用量虽少,但杂质含量会增加,茶多酚的提取率降低。通过试验,本实验采用的酸试剂为0.5 mol/L~1.0 mol/L的HCl,结果见图1。

图1 酸的浓度在茶多酚提取中的影响Fig.1 Effect of acid concentration on the extraction of TP
1.3.4 乙酸乙酯萃取试验
乙酸乙酯萃取试验结果见表2。
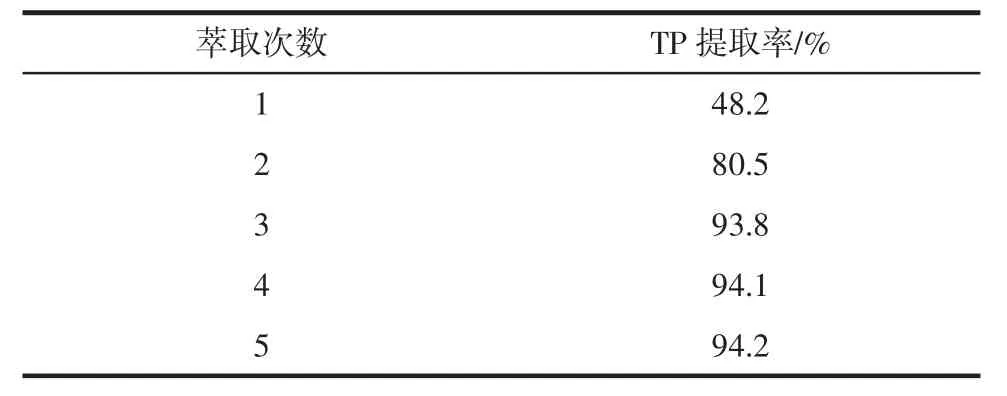
表2 萃取次数对茶多酚提取率的影响Table 2 Effect of extraction times on the extraction of TP
如表2所示,当萃取次数大于或等于3次后,茶多酚的提取率变化不大。所以为了节省工序,沉淀转溶后用乙酸乙酯萃取三次即可,每隔5分钟萃取一次。萃取时间也不宜过长,因为在酸性条件下,乙酸乙酯易水解[7]。
1.3.5 减压蒸馏试验
将乙酸乙酯萃取后的有机相合并,减压蒸馏浓缩,减压温度不宜过高,以55℃为宜,浓缩液真空干燥后冷藏,得样品Ⅰ。
1.3.6 硅胶柱层析实验
将200目~300目硅胶用石油醚湿法装柱,样品Ⅰ上样,用事先配好的洗脱剂(本实验选用的洗脱剂是石油醚、乙酸乙酯和甲酸按不同比例配成极性由小到大的溶液)洗脱,极性逐渐加大,每30毫升接一次溶液,减压蒸馏,收集样品Ⅱ。按色带的不同先后收集了三部分,分别标记为 T1、T2,为浅褐色粉末状;T3、T4、T5、T6、T7、T8、T9、T10,为白色针状晶体;T11、T12、T13,为浅黄色晶体[8-9]。
2 分析方法
2.1 福林酚试剂比色法分光光度分析方法
2.1.1 原理
利用福林酚(Folin-Ciocalteu)试剂氧化茶多酚中的-OH基团会显蓝色的性质,用分光光度计测茶多酚的含量。最大吸收波长λmax为765 nm,用没食子酸作校正标准定量茶多酚[10]。
2.1.2 结果与分析
根据GBIT 8313-2008《茶叶中茶多酚和儿茶素类含量的检测方法》,称取茶多酚粗品0.2 g,样品T11为0.03 g,稀释一定倍数后分别装入容量瓶1号,2号中。

式中:A为吸光值;稀释因子d分别为100、10;样品干物质含量m=100%;样品质量m1=0.2 g,0.03 g;V=10mL;SLOPEStd为没食子酸标准曲线的斜率1.1159。根据上面公式绘制表3。

表3 茶多酚粗品和茶多酚精制品的含量Table 3 Effect of extraction times on the extraction of TP
如表3所示,茶多酚粗品和样品T11中茶多酚的含量。因为茶多酚主要包括儿茶素类、黄酮类、花色素类和酚酸类四种物质,所以该数据是指这四种物质的总含量。
2.2 高效液相色谱分析方法[11-18]
2.2.1 仪器与试剂
Dionex分析型高效液相色谱仪(Ultimate 3000);KQ-250DE数控超声波清洗器:昆山市超声仪器有限公司;UPT-II-20T优普超纯水机:成都超纯科技有限公司。
甲醇,乙酸,乙腈(色谱纯):山东禹王试业有限公司化工分公司;标品 GA,C,CAF,EC,EGCG,GCG 和ECG,纯度≥99%(阿拉丁公司产品)。
2.2.2 高效液相色谱条件
色谱柱:AT LiCHPOM C18(250 mm×4.6 mm,5 μm)。检测波长:278nm。柱温:35℃。流速:1.000 mL/min。进样量:50μL。A流动相的配制:90mL乙腈加5mL乙酸定容于1000mL容量瓶,震荡摇匀,过有机膜(0.45μm),超声脱气;B流动相的配制:400mL乙腈加2.5mL乙酸定容于500mL容量瓶,震荡摇匀,过有机膜(0.45μm),超声脱气。梯度洗脱条件:0min~10min,100.0%A;10min~25 min,100.0%~68.0%A,0.0%~32.0%B;25 min~30 min,68.0%A,32.0%B;30 min~35 min,68.0%~100.0%A,32.0%~0.0%B;35min~40min,100.0%A,0.0%B。
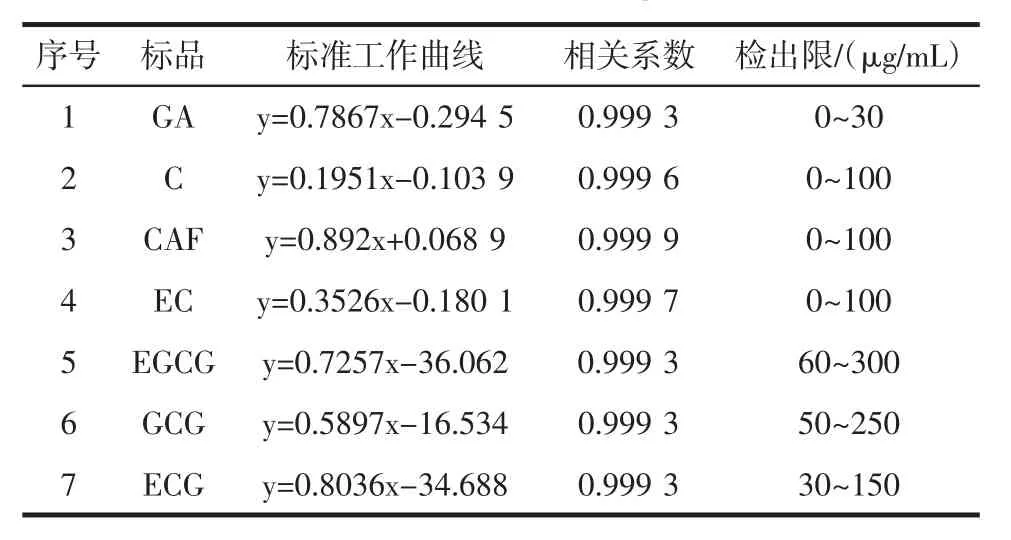
表4 标准工作曲线Table 4 Standard working curve
2.2.3 高效液相色谱分析方法
2.2.3.1 稳定溶液的配制方法
将50 mL乙腈加入500 mL容量瓶中,用水定容至刻度,摇匀后过有机膜(0.45 μm),超声脱气。
2.2.3.2 标准工作曲线
根据GB/T 8313-2008《茶叶中茶多酚和儿茶素类含量的检测方法》,将标品稀释不同浓度后,分别测出相应浓度下各标品的保留时间(RT)和积峰面积,作出标品积峰面积(mAU)—浓度(μg/mL)曲线,通过标准曲线计算样品各成分含量,结果见表4。如图2所示,原料茶多酚粗品中GA、C、CAF、EC的含量较少,所以这四条工作曲线的线性范围从0 μg/mL开始。
2.2.3.3 定性分析
将茶多酚粗品和试验制得的13个样品(T1~T13)用稳定溶液稀释一定倍数后,在2.2.2的色谱条件下,所得的高效液相图谱,通过与标准品图谱的比较,确定出样品中各生理活性成分峰,结果见图3。
2.2.3.4 定量分析
根据标准品标准曲线方程如表4所示,计算样品中各成分的浓度X(μg/mL),再代入如下公式计算出样品中各成分的含量,结果如表5所示。

式中:M为样品中各成分的含量,%;V为样品定容体积,mL;m 为样品质量,μg;d为稀释倍数。
2.2.4 结果与分析
如表5所示,通过计算得到了样品中各主要成分的百分含量;T1和T2除了含没食子酸、咖啡碱较多外,HPLC谱图中还存在很多杂质峰;T3~T10的HPLC谱中只有 GA 峰(RT≈5.45)和 CAF峰(RT≈18.990),且可看到没食子酸的含量逐渐减少,而咖啡碱的含量逐渐增加后又逐渐减少;T11~T13的没食子酸含量保持在3.00%左右,咖啡碱含量保持在2.00%左右。
3 结论
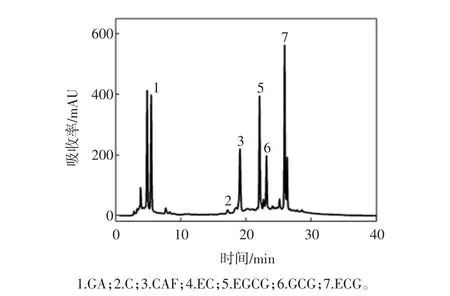
图2 茶多酚粗品HPLC图谱Fig.2 HPLC of tea polyphenol
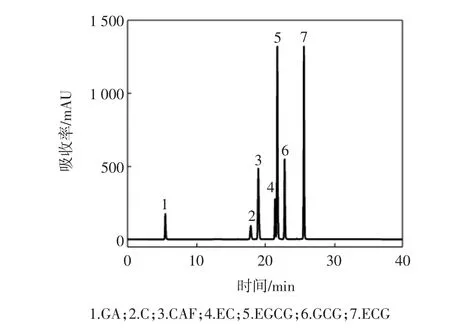
图3 混标溶液HPLC图谱Fig.3 HPLC of standard specimen

表5 样品中各成分含量Table 5 Contents of components in samples%
本试验结合金属离子沉淀法和硅胶柱层析法的优点,尝试了沉淀-柱层析试验。通过福林酚试剂比色法和高效液相色谱法,由分析数据可知,先用沉淀法去除茶多酚粗品中的咖啡碱,再用硅胶柱层析法精制茶多酚的方法是可行的。
该方法提取茶多酚的最佳工艺条件为:温度50℃下用60%乙醇水溶液溶解茶多酚粗品,转速40 r/min,复合沉淀剂 m(AlCl3)∶m(ZnCl2)=1 ∶2,沉淀条件为1.0 mol/LNaHCO3调节 pH=5.5~6.0,0.5~1.0 mol/LHCl转溶沉淀,乙酸乙酯萃取三次,减压蒸馏温度为55℃,柱层析法的洗脱剂选择石油醚,乙酸乙酯和甲酸按比例配制的溶液。
比较表3和表5,酮类、花色素类和酚酸类这三种物质在茶多酚粗品和样品T11中的含量分别为3.89%、0.4%。说明沉淀-柱层析法还可以有效地除去粗品中的黄酮类、花色素类和酚酸类物质。如表5所示,T11~T13中C和EC的含量较少,EGCG的含量最多;儿茶素的总量从粗品的20.34%到T11的80.70%,杂质明显减少,含量也有显著增高。进一步印证了沉淀-柱层析法精制茶多酚的方法是可行的。遗憾的是样品中没食子酸和咖啡碱从始至终都存在,而且由于儿茶素各单体在有机溶剂中的极性相近,硅胶柱层析法无法将其很好的分开。
:
[1]Yang C S,Landau J M.Effect s of tea consumption on nutrition and health[J].Journal of nutrition,2000,130(10):2409-2412
[2]刘世初,孙志洪,王斌.茶多酚的提取工艺及其应用机理研究进展[J].家畜生态学报,2009,30(4):91-94
[3]蒋建平,陈洪,汪秋安,等.茶多酚的离子沉淀法提取及其成分分析[J].株洲工学院学报,2004,18(5):53-56
[4]余兆祥,王筱平.复合型沉淀剂提取茶多酚的研究[J].食品工业科技,2001,22(3):32-34
[5]邓泽元,刘娟.金属盐沉淀法提取茶多酚影响因素的研究[J].南 昌大学学报,2001,23(3):92-97
[6]周志,汪兴平,莫开菊,等.Al3+沉淀法对茶多酚制备效果的影响研 究[J].食品科学,2002,23(6):88-90
[7]马红青,王朝瑾.溶剂法萃取茶多酚的工艺研究[J].食品科学,2008,29(11):179-182
[8]朱斌,陈晓光.二次柱层析制备高纯度表没食子儿茶素没食子酸 酯(EGCG)的工艺研究[J].食品与机械,2009,25(4):83-85
[9]袁华,吴莉,吴元欣,等.硅胶柱层析法提纯茶多酚的研究[J].华中 师范大学学报:自然科学版,2007,41(4):554-556
[10]孙健,李楠,邢国秀,等.茶多酚高效液相指纹谱-分光光度法质量 分析研究[J].食品科学,2001,22(11):63-65
[11]莫燕霞,胡宝祥,胡伟,等.高效液相色谱法测定茶叶中茶多酚[J].理化检验:化学分册,2008,44(7):593-596
[12]戴军,王洪新,陈尚卫,等.茶叶及茶多酚中儿茶素的高效液相色 谱分析方法研究[J].色谱,2001,19(5):398-402
[13]Wang H F,Gordon J P,Keith H.HPLC determination of catechins in tea leaves and tea extracts using relative response factors[J].Food chemistry,2003,81(2):307-312
[14]Pelillo M,Biguzzi B,Bendini A,et al.Preliminary investigation into development of HPLC with UV and MS-electrospray detection for the analysis of tea catechins[J].Food chemistry,2002,78(3):369-374
[15]Bronner W E,Beecher G R.Method for determining the content of catechins in tea infusions by high-performance liquid chromatography[J].Journal of chromatogr A,1998,805(1):137-142
[16]Finger A,Kuhr S,Engelhardt U H.Chromatography of tea constituents[J].Journal of Chromatogr A,1992,624(1):293-315
[17]Goto T,Yoshida Y,Kiso M,et al.Simultaneous analysis of individual catechins and caffeine in green tea[J].Journal of Chromatogr A,1996,749(1):295-299
[18]Amarowicz R,Shahidi F.A rapid chromatographic method for separation of individual catechins from green tea[J].Food research international,1996,29(1):71-76
