一个含苯甲酸配体的Mn-Ce 混合金属簇合物的合成、晶体结构与磁性研究
2013-08-20王会生潘志权
王会生 潘志权
(绿色化工过程教育部重点实验室,武汉工程大学化工与制药学院,武汉 430073)
自从1993 年以来,含MnⅢ离子的高核Mn 簇合物单分子磁体引起国内外化学家、物理学家和材料学家的极大的兴趣,主要源于这类材料在高密度信息存储设备、 量子计算机等方面存在潜在应用[1-4]。目前,合成含有MnⅢ高核Mn 簇合物的方法主要有MnO4-离子与Mn2+的反歧化反应[5]、KMnO4或NBut4MnO4(But为正丁基)在含有羧酸配体的甲醇溶液中进行的还原聚合反应[6]、Mn2+离子在碱性溶液中进行的氧化反应[7]、三核(四核或其它核数)的高核Mn 簇合物前驱体在含有各种螯合配体的有机溶液中进行的反应[8]。此外,还有一种合成含MnⅢ离子的Mn 或Mn-Ce 混金属簇合物的简易方法是使用CeⅣ作为氧化剂氧化Mn2+进行的反应,截至目前,已通过这种方法合成出[Mn2Ce][9]、[Mn2Ce3][10]、[Mn6Ce][11]、[Mn4Ce2][12]、[Mn8Ce][13]、[Mn10Ce4][14]等不同结构构型的Mn-Ce 混合金属簇合物。王会生等也通过这种方法合成出一系列马鞍形骨架[CeⅣMnⅢ8O8]12+簇合物:[CeⅣMnⅢ8O8(O2CMe)12(H2O)4]·4H2O,[CeⅣMnⅢ8O8(O2CMe)12(py)3(H2O)]·6.5H2O 及[CeⅣMnⅢ8O8(O2CMe)12(bzd)2(H2O)2]·10H2O(其中py 为吡啶配体;bzd 为苯并咪唑配体)[15]。值得一提的是,上述这些不同核结构的Mn-Ce 簇合物所使用的合成方法都是Ce4+氧化溶解在H2O/CH3CO2H 中的Mn2+离子,因此得到的都是含醋酸配体的化合物。到目前为止,还没有通过这种方法得到含苯甲酸配体的Mn-Ce 簇合物。由于反应非常敏感的受到溶剂的质子性、 极性等的影响,我们在本工作中使用乙腈-甲醇混合溶剂,使用不含醋酸配体的Mn(O2CPh)2·2H2O,然后用(NH4)2Ce(NO3)6氧化其中的Mn2+,获得一个结构新颖的含苯甲酸配体的五核Mn-Ce 混合金属簇合物[Mn3Ce2(O)5(O2CPh)9(CH3OH)3]·2CH3CN(1·2CH3CN),X-射 线单晶结构分析表明其中2 个Mn 原子、2 个Ce 原子及4 个桥连O 原子构成不规则的立方烷,然后通过1 个桥连O 原子连接到另1 个Mn 原子上。磁性研究表明分子内Mn 离子之间存在铁磁相互作用,该化合物的基态自旋值S=5,D=-0.31 cm-1,但交流磁化率测试没有发现频率依赖现象。
1 实验部分
1.1 试剂和仪器
所有药品和化学试剂均为分析纯,使用时没有进行进一步提纯。Mn(O2CPh)2·2H2O 按文献方法合成[16]。元素C、H 分析使用Perkin-Elmer 240C 元素分析仪测定。红外光谱使用VECTOR 22 光谱仪,并用KBr 压片法测定,测量范围为400~4 000 cm-1。磁性采用多晶样品,在MPMS-XL7 SQUID 磁测量仪上测定,直流磁化率的测量温度为1.8~300 K,测量磁场为2 000 Oe,抗磁部分用Pascal 常数校正[17];磁化测量温度为1.8~10.0 K,测量磁场为1.0~7.0 T;交流磁 化 率 的 测 量 频 率 为1、10、100、498、997、1 488 Hz,振荡场为3.0 Oe,直流场为0 Oe。
1.2 化合物的合成
Mn(O2CPh)2·2H2O(0.25 g,0.75 mmol)和 苯 甲 酸(0.274 8 g,2.25 mmol)溶解在由10 mL 乙腈和5 mL甲醇组成的混合溶剂中,搅拌,然后逐份加入(NH4)2Ce(NO3)6(0.205 6 g,0.375 mmol),加完后,继续搅拌30 min,过滤,滤液为深红色,空气中静置约5 d,析出红色晶体,过滤,用冷MeCN 和冷Et2O 各洗涤三次,空气干燥,产率约为60%(based on Mn)。元素分析:按分子式C66H57Ce2Mn3O26(2 个乙腈溶剂分子已失 去),计 算 值(%):C 46.32,H 3.36;实 测 值(%):C 46.33,H 3.42。红外光 谱IR (KBr,cm-1):3 060(w),1 689(m),1 600(s),1 535(s),1 448(m),1 384(s),1 174(m),1 024(m),920(m),842(m),715(s),622(s),560(s),472(s),433(s)。
1.3 单晶结构的测定
单晶结构分析用Bruker SMART APEX CCD 单晶衍射仪,Mo Kα 辐射(λ=0.071 073 nm)及φ-ω 扫描方式,采用SMART 程序在室温下收集衍射数据。运用SAINT 程序还原数据和SADABS 程序进行经验吸收校正[18-19]。结构用直接法解出[20],并基于F2用全矩阵最小二乘法对结构进行精修。非氢原子采用各向异性热参数精修。所有的氢原子均为理论加氢并采用各向同性热参数及跨式模型 进行修正,且Uiso是母原子的1.2 或1.5 倍。化合物1·2CH3CN 的部分晶体数据和结构精修参数列于表1 中,相应的重要键长键角列于表2 中。
CCDC:912993。
2 结果与讨论
2.1 晶体结构
单晶X-射线分析表明,化合物1·2CH3CN 属于三斜晶系空间群,晶体结构图如图1a 所示,核结构如图1b 所示。该化合物由1 个 [Mn3Ce2(O)5(O2CPh)9(CH3OH)3]异核金属簇合物和2 个乙腈分子组成,其中簇合物由3 个Mn 原子(Mn1、Mn2 和Mn3)和2 个Ce 原子(Ce1 和Ce2)组成,这些金属原子被5 个μ3-O 连接而成。其中2 个Ce 原子、2 个Mn 原子和4 个μ3-O(O19、O20、O21 和O22)组成1个不规则的立方烷,然后再通过1 个μ3-O(O23)连接到Mn3 上。此外,据作者所知,这种核结构在Mn-Ce混合金属簇合物中第一次出现。外围配体由9 个苯甲酸根配体和3 个端基配位的甲醇配位分子提供,其中9 个苯甲酸根配体都是常见的的μ2-配位模式。
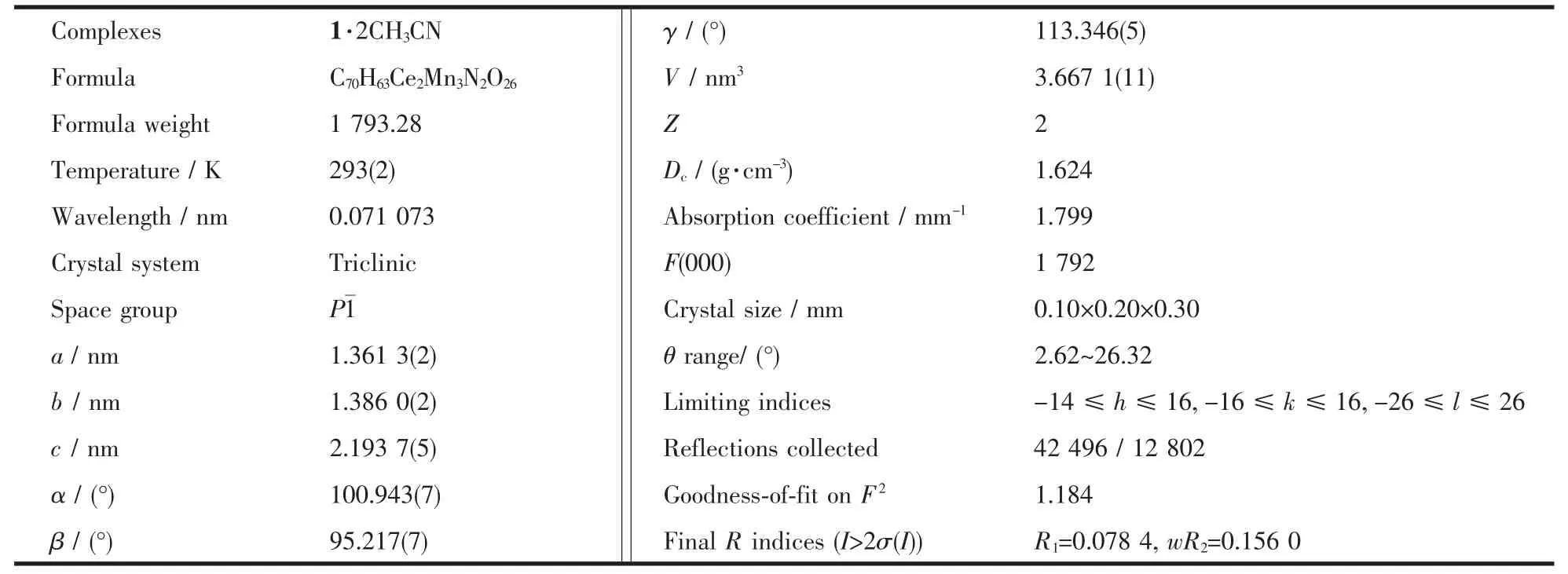
表1 化合物1·2CH3CN 的部分晶体数据和结构修正参数Table 1 Summary of the crystal data and the refinement details for complex 1·2CH3CN
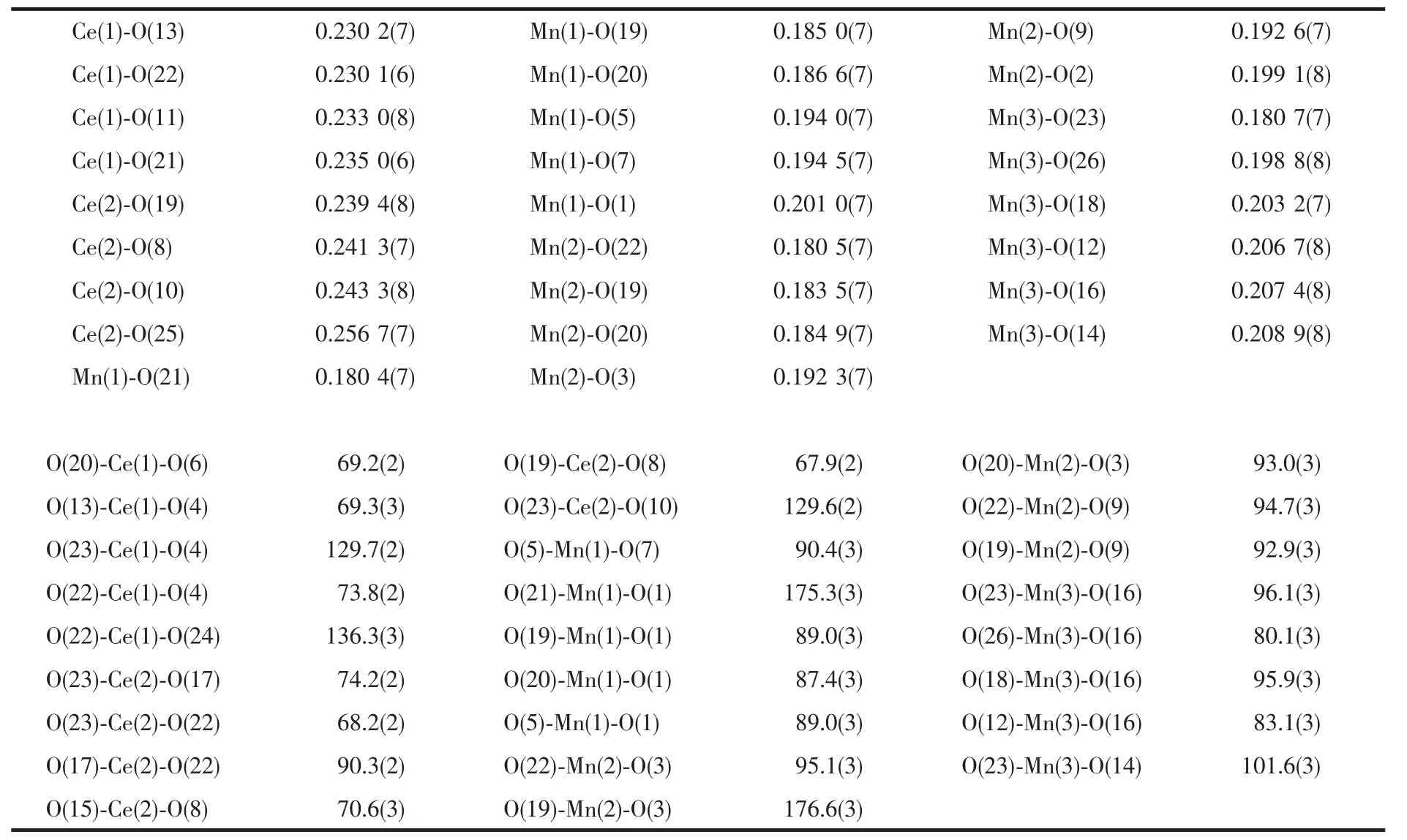
表2 化合物1·2CH3CN 的部分键长键角Table 2 Selected bond lengths (nm) and bond angles (°) for complex 1·2CH3CN
该化合物中金属离子Mn 和Ce 的价态和O 原子的质子化水平通过电荷平衡、仔细检查相应的键参数及价键和计算确认[21-23],结果(如表3 所示)表明Mn1 和Mn2 为+4 价,Ce1 和Ce2 也 为+4 价,Mn3为+3 价,5 个μ3-O 原子O19、O20、O21、O22 和O23为O2-离子,甲醇分子都没失去质子且苯甲酸配体都是脱质子的。化合物中所有Mn 都是六配位八面体构型,对于Mn1 和Mn2 的Mn-O 键长范围为0.180 4~0.201 0 nm,其键长值符合MnⅣ的Mn-O 键长范围,其次通过没有明显的Jahn-Teller 轴也说明这2 个离子为+4 价;而对于Mn3 原子同其它含MnⅢ离子的化合物一样,也有Jahn-Teller 效应,但表现出非常少见的压扁八面体结构,其缩短轴为O23-Mn3-O26,键长Mn3-O23 0.180 7 nm 和Mn3-O26 0.198 8 nm 比 该Mn 原 子 上 其 它4 个 键 长 都 短(Mn3-O16 0.207 4 nm,Mn3-O18 0.203 2 nm,Mn3-O12 0.206 7 nm,Mn3-O14 0.208 9 nm)。化 合物中Ce 为九配位三帽三棱柱结构,Ce-O 键长范围为0.227 4~0.256 7 nm,其值为Ce4+离子的Ce-O 键长特征。
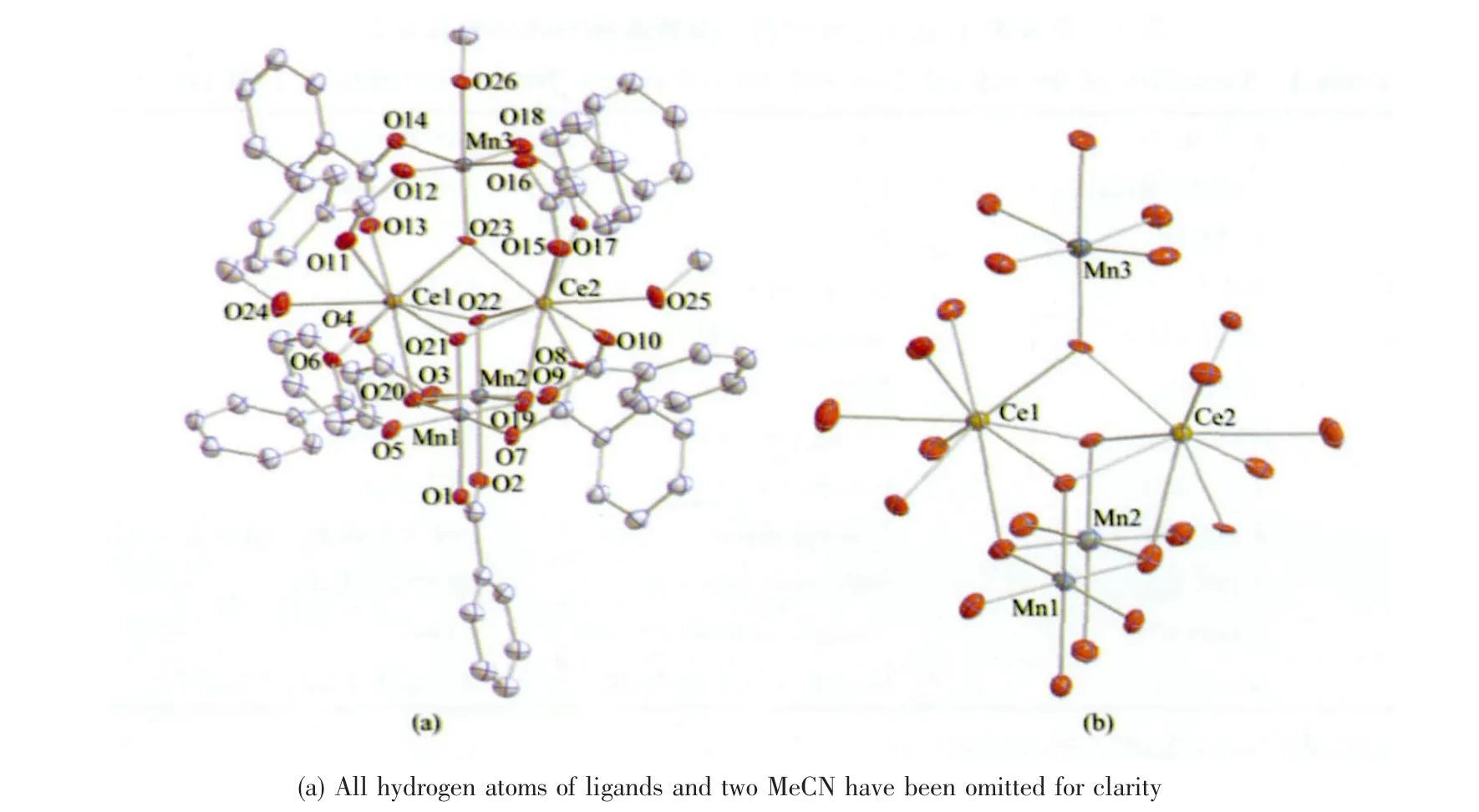
图1 (a) 簇合物1 的晶体结构, (b) 簇合物1 的核骨架Fig.1 (a) Crystal strucuture of complex 1 and (b) core of complex 1 with 30% probability level

表3 化合物1·2CH3CN 中Mn 和Ce 的价键和计算Table 3 Bond valence suma for the Mn and Ce atoms of 1·2CH3CN
簇合物中立方烷内MnIV-MnIV之间的距离为0.270 6 nm,CeⅣ-CeⅣ之间的距离为0.347 0 nm,MnⅣ-CeⅣ之间的距离范围为0.326 5~0.329 0 nm;连接在立方烷外的另1 个MnⅢ原子与2 个CeⅣ之间的距离分别为0.371 6 nm 和0.374 6 nm。由这些金属原子之间的距离可见立方烷内2 个MnⅣ离子之间磁耦合作用较强,但由于CeⅣ离子的f 轨道上电子数为0,不与和它相连的MnⅣ或MnⅢ发生磁耦合作用,但可作为介质使得MnⅣ和MnⅢ之间发生较弱的磁耦合作用。在晶胞内,不同簇合物上Ce…Ce 之间的最短距离为1.154 8 nm,Mn…Mn 最短距离为0.955 6 nm,Mn…Ce 最短距离为1.041 7 nm,此外,除了晶格内不同簇合物苯甲酸上的苯环之间存在一定强度的π-π 堆积作用(π 环的质心距离为0.366 5 nm,如图2 所示)外,没有分子间氢键存在。总之,分子间除π-π 堆积造成的分子间作用外,其它分子间作用比较微弱。
2.2 磁学性质
化合物1 的变温磁化率曲线如图3 所示。在300 K 时,化合物1 的χMT 值为7.32 cm3·K·mol-1,其 值 比2 个Mn4+离 子、1 个Mn3+离 子χMT 值6.75 cm3·K·mol-1高了一些(CeⅣ的f 轨道电子数为0,抗磁性的)[24]。当温度降低时,χMT 值逐渐升高,到约为8.0 K 时,χMT 达到最大值14.34 cm3·K·mol-1,然后随着温度进一步降低,χMT 急剧下降,到1.8 K 时,其值为12.91 cm3·K·mol-1。直流磁化率曲线随着温度降低而升高,说明该化合物中离子间是铁磁性耦合的。由于CeⅣ是抗磁性的,因此可以假设Mn1、Mn2 之间的耦合常数为J1、Mn1 与Mn3 之间和Mn2与Mn3 之间的耦合常数都为J2,然后使用MAGPACK 软件进行拟合[25],拟合结果为:J1=12.45 cm-1,J2=1.95 cm-1,g=1.99,R=9.74×10-5,拟合结果也说明Mn4+之间铁磁性耦合确实比Mn4+与Mn3+之间的大了许多,这与结构分析中磁耦合相对强弱的判断一致。需要说明的是,低于8.0 K 的χMT 急剧下降主要由于分子间弱的反铁磁相互作用、Zeeman 效应和/或零场分裂作用导致的。
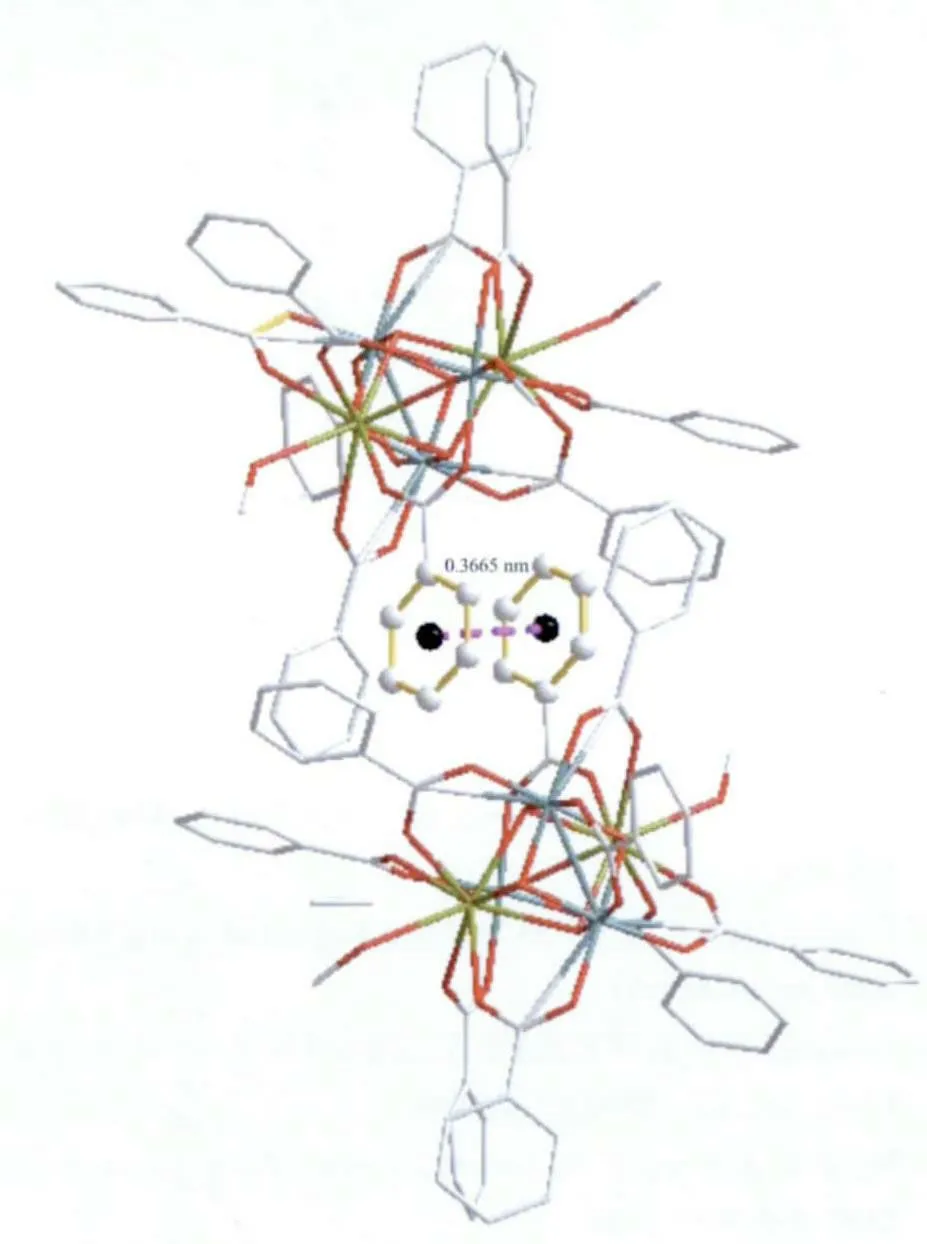
图2 化合物1 晶体中的π-π 堆积作用Fig.2 π-π stacking interactions in the crystal for 1
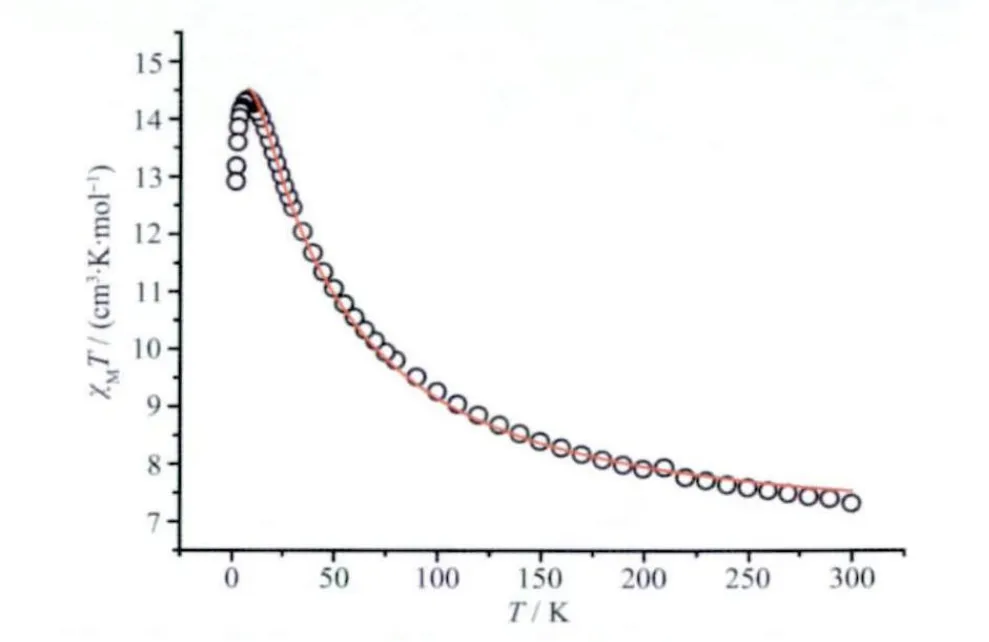
图3 化合物1 的变温磁化率曲线Fig.3 Temperature dependence of χMT for 1
为了确定该1 的基态自旋值S 和各向异性参数D,化合物的磁化曲线在2.0~7.0 T 的磁场、1.8~10.0 K 的条件下进行测量,其(M/N)μB对H/T 图和M/(NμB)对H 图分别如图4a 和4b 所示。其7 T 的磁化最大值为9.59μB。不同磁场时每条磁化曲线不重合,说明存在磁各向异性,其可由图4b 所示的7 T时磁化还没饱和可以看出来。根据7 T 时的磁化强度为9.59μB,对应基态自旋S=5,则用ANISOFIT 软件拟合[26],可得到拟合效果很好的曲线(如图4a),得到的参数为:g=1.97,S=5,D=-0.31 cm-1。
为了进一步研究化合物1 是否有单分子磁体性质,我们对该化合物进行交流磁化率测试,其交流磁化率的实部( χM′)和虚部( χM″)随温度变化曲线如图5a 和5b 所示。由图可知,该化合物的实部和虚部在温度高于1.8 K,频率达到1 488 Hz 时都没有出现频率依赖现象,说明在我们测量条件下并没有慢弛豫行为,这可能由于化合物的能垒比较低有关。
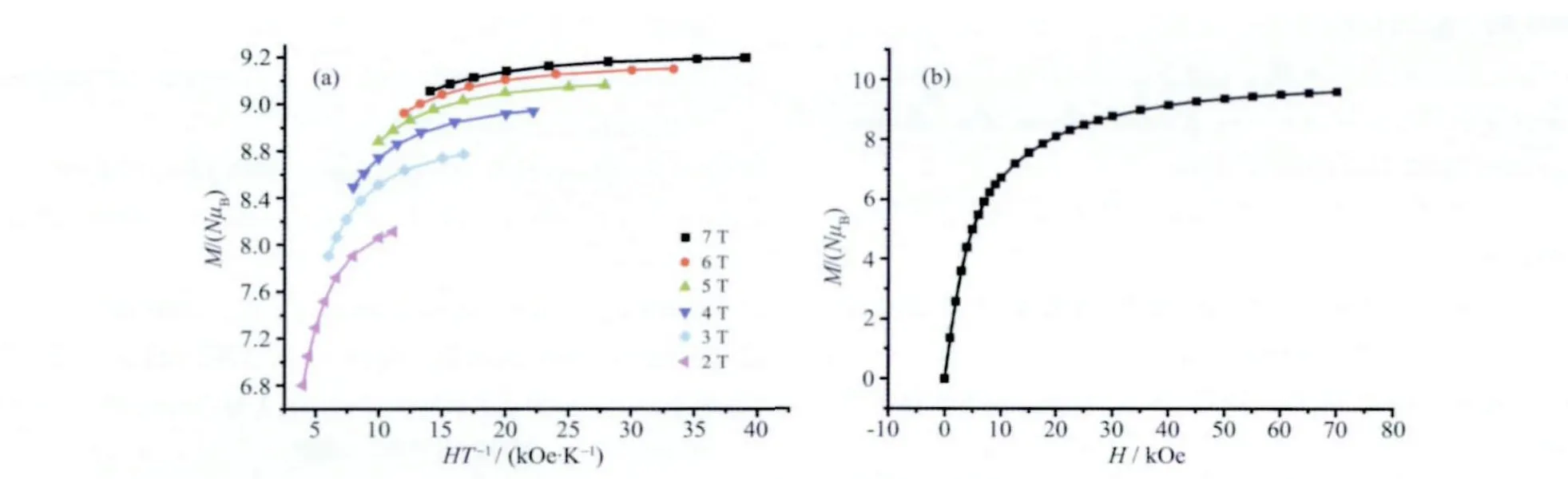
图4 (a) 化合物1 的(M/N)μB 对H/T 曲线,图中实线代表用ANISOFIT 软件拟合出的数据;(b) 1.8 K 时M/(NμB)对H 曲线Fig.4 (a) Plot of M/(NμB) vs H/T for 1, the solid lines represent the fitted result; (b) Plot of M/(NμB) vs H at 1.8 K for 1
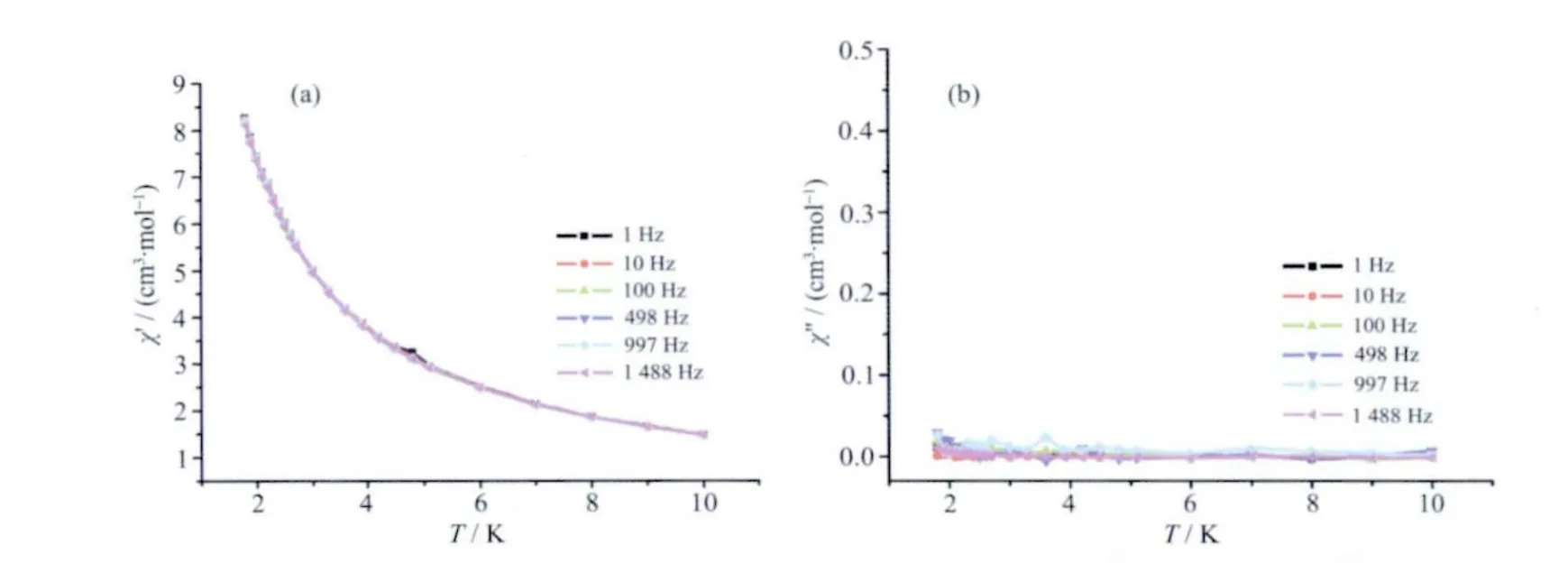
图5 化合物1 的交流磁化率实部( χM′, a)和虚部( χM″, b)随温度变化曲线,所测频率如该图中内图所示Fig.5 Plot of the temperature dependence of the in-phase ( χM′, a) and out-of-phase ( χM″, b)AC susceptibility signals for complex 1 at the indicated frequencies
3 结 论
在本工作中,我们把苯甲酸配体、Mn(O2CPh)2·2H2O 溶解在甲醇-乙腈混合溶剂中,并用(NH4)2Ce(NO3)6氧化溶液中的Mn2+,获得一个结构新颖的[Mn3Ce2]混合金属簇合物,并对其进行单晶结构分析、红外光谱、元素分析及磁性表征。磁性研究表明,簇合物内Mn4+之间的磁耦合作用J1=12.45 cm-1,Mn4+与Mn3+的磁耦合作用J2=1.95 cm-1,基态自旋值和磁各向异性参数分别为5 和-0.31 cm-1,但没有发现交流磁化率虚部的频率依赖现象,可能由于能垒较低造成的。
[1] Aromí G, Brechin E K. Struct. Bonding(Berlin), 2006,122:1-67
[2] Gatteschi D, Sessoli R. Angew. Chem. Int. Ed., 2003,42:268-297
[3] Wernsdorfer W, Soler M, Christou G, et al. J. Appl. Phys.,2002,91:7164-7166
[4] WANG Tian-Wei(王天维), LIN Xiao-Ju(林小驹), WEI Ji-Zong(韦 吉 宗), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2002,18(11):1071-1080
[5] Wemple M W, Tsai H L, Claude J P, et al. Inorg. Chem.,1996,35:6437-6449
[6] Tasiopoulos A J, Wernsdorfer W, Abboud K A, et al. Angew.Chem. Int. Ed., 2004,43:6338-6342
[7] Liu C M, Zhang D Q, Zhu D B. Inorg. Chem., 2009,48:792-794
[8] Ma Y S, Song Y, Li Y Z, et al. Inorg. Chem., 2007,46:5459-5461
[9] Ramalakshmi D, Rajasekharan M V. Acta Cryst., 1999,B55:186-191
[10]Tasiopoulos A J, Jr P L M, Abboud K A, et al. Inorg. Chem.,2007,46:9678-9691
[11]Tasiopoulos A J, O′Brein T A, Abboud K A, et al. Angew.Chem. Int. Ed., 2004,43:345-349
[12]Milios C J, Wood P A, Parsons S, et al. Inorg. Chim. Acta,2007,360:3932-3940
[13]Mishra A, Tasiopoulos A J, Wernsdorfer W, et al. Inorg.Chem., 2008,47:4832-4843
[14]Mishra A, Tasiopoulos A J, Wernsdorfer W, et al. Inorg.Chem., 2007,46:3105-3115
[15]Wang H S, Ma C B, Wang M, et al. J. Mol. Struct., 2008,875:288-294
[16]Wemple M W, Tsai H L, Wang S, et al. Inorg. Chem., 1996,35:6437-6449
[17]Canty A J, Minchin N J, Patrick J M, et al. Dalton Trans.,1983:1253-1259
[18]SAINT-Plus, Version 6.02; Bruker Analytical X-ray System:Madison, WI, 1999.
[19]Sheldrick G M.SADABS:An Empirical Absorption Correction Program; Bruker Analytica X-ray Systems, Madison, WI,1996.
[20]Sheldrick G M. SHELXTL-97; Universität of Göttingen,Göttingen, Germany, 1997.
[21]Liu W, Thorp H H. Inorg. Chem., 1993,32:4102-4105
[22]Brown I D, Shannon R D. Acta Crystallogr., 1973,A29:266-282
[23]Roulhac P L, Palenik G J. Inorg. Chem., 2003,42:118-121
[24]Benelli C, Gatteschi D. Chem. Rev., 2002,102:2369-2387
[25]Borras-Almenar J J, Clemente-Juan J M, Coronado E, et al.Inorg. Chem., 1999,38:6081-6088
[26]Shores M P, Sokol J J, Long J R. J. Am. Chem. Soc., 2002,124:2279-2292
