粒度可调纳米CuO 的制备及其表面硫化研究
2013-08-20顾少楠孙和云范迎菊孙中溪
顾少楠 孙和云 范迎菊 孙中溪
(济南大学化学化工学院,济南 250022)
不同形貌的氧化铜和表面改性氧化铜纳米粒子在气敏传感[1-8]、催化[9-15]、电化学传感器[16-18]等方面表现出不同寻常的特性,因此其制备方法和性能一直是近些年的研究热点[19-30]。采用传统的直接沉淀法制备纳米氧化铜粉体,虽然有操作方便、设备简单、成本较低等特点,但产物粒度较大,不利于纳米氧化铜粉体特性的发挥。如何有效简便的控制纳米氧化铜的粒度并对其进行表面改性一直是一个富有挑战性的研究课题。
铜金属的工业矿物主要有硫化铜矿和氧化铜矿两大类。而铜的硫化矿物可选性显然优于相应的氧化矿物。随着易选硫化铜矿产资源的日趋枯竭,难选的氧化铜矿的分离浮选成为重要的研究课题。由大量的理论研究和工艺实践证明,对氧化铜矿物表面进行硫化后再进行浮选是一种有效的选矿工艺。
为使贫矿和复杂矿中有用矿物实现单体解离,长时间磨矿过程是必不可少的。而这个磨矿过程,往往会产生大量难以分选的超细粒氧化铜矿物。为提高细粒氧化铜矿的分选效率就有必要深入了解氧化铜矿的表面化学性质和表面硫化机理,这对于提高超细粒氧化铜矿的分选效率,从而解决铜矿资源的匮乏,保持国民经济的可持续发展具有重要意义。为了解氧化铜的硫化机理,我们采用人工合成氧化铜矿物作为研究样品以排除来自不同矿山纯矿物的代表性不具普适性的不足和矿物杂质的影响。
本文报告一种如何在醇-氨水混合体系中制备纳米氧化铜粉体并采用热处理方法对合成的氧化铜纳米粒子实现表面硫化改性的简单方法。通过利用氮气吸附/脱附(BET)、粉末X 射线衍射(XRD)、红外光谱(IR)等手段对合成产品的表征,证明本方法能够制备出粒径大小可调的氧化铜纳米粒子,并实现对氧化铜纳米粒子的表面硫化。本研究不仅对于了解细粒氧化铜矿物的表面硫化浮选机理有重要意义,也可为核壳结构纳米材料的制备和表面改性研究所借鉴。
1 实验部分
实验用水均为二次蒸馏水,实验所用的乙基黄原酸钾由实验室制备和提纯,纯度99%以上,其它试剂均为分析纯,由国药集团化学试剂有限公司等国内试剂厂家提供。
1.1 纳米氧化铜的制备及表面硫化
称取1.88 g Cu(NO3)2·3H2O,配制0.10 mol·L-1硝酸铜溶液并在100 mL 容量瓶中定容; 称取1.2 g NaOH,在100 mL 烧杯中用20 mL 蒸馏水溶解,再在100 mL 容量瓶中用无水乙醇定容,配制0.3 mol·L-1NaOH 溶液。将配制的铜离子溶液移入烧杯中并在磁力搅拌器不断搅拌下缓慢加入25%氨水约25 mL,配位后以约0.1 mL·d-1、1~2 d·s-1的速度分别滴加100 mL NaOH 溶液。将所得沉淀减压抽滤,反复洗涤3 次,然后在温度为65 ℃条件下真空干燥2 h。将干燥后样品在温度为370 ℃的马弗炉中煅烧2 h,得到黑色纳米CuO 粉体。
按上述顺序和操作进行实验,只是将无水乙醇分别换用正丁醇和正辛醇,同样得到黑色纳米CuO粉体。
分 别 取0.25 g CuO(比 表 面 为9.88 m2·g-1)和0.003 2 g 单质升华硫,在玛瑙研钵内均匀研细,放入瓷舟中,在温度为200 ℃的管式炉氮气保护下煅烧90 mins 得到表面硫化的CuO(CuO/CuS)。
1.2 样品的表征
所制备的产物物相均用D/max-γA 型旋转阳极X 射线衍射仪进行测定,X-射线源为Cu Kα 辐射(λ=0.154 18 nm),扫描角度范围2θ 为10°~80°。热重-差热分析采用美国珀金埃尔默公司Diamond TG/DTA 型热重分析仪,升温速率10 ℃·min-1,气氛为空气,空气流速20.0 mL·min-1。比表面积测定采用美国Quantachrome 公司Nova2000e 系列比表面积及孔径分析仪,选定氮气为吸附气体,测定温度为液氮温度(77 K),在此范围自动选择测定样品点数。样品在脱气站的脱气温度为150 ℃,脱气时间为6 h。红外光谱采用美国PE 公司生产的SPECTRUM ONE 傅立叶变换红外光谱仪进行测试,KBr 压片,波 长 扫 描 范 围400 ~4 000 cm-1。使 用 日 本SHIMADZU 公司的UV-2450 型紫外可见分光光度计,通过测定残余烷基黄原酸盐的吸光度来确定其吸附量。
1.3 标准乙基黄原酸钾溶液的配制及吸附实验
准确称取0.080 2 g 乙基黄原酸钾,溶于少量去离子水,移入到500 mL 容量瓶中,准确定容至刻度,配制出浓度为1 mmol·L-1的乙基黄原酸钾溶液,再用移液管从配好的溶液中准确移取10 mL 溶液于另一个100 mL 的容量瓶中,稀释10 倍得到0.1 mmol·L-1的乙基黄原酸钾溶液。
分 别 准 确 移 取0、5、10、15、20、25 mL 的0.1 mmol·L-1的乙基黄原酸钾溶液于6 个25 mL 的容量瓶中,分别准确定容至刻度,用紫外可见光谱仪做出乙基黄原酸钾的标准曲线,最大吸收波长为301 nm。
分别称取0.01 g CuO 和CuO/CuS 置 于50 mL离心管,向离心管中加入25 mL 浓度为1 mmol·L-1的乙基黄原酸钾溶液,调节pH 为10.5 左右,振荡2 h 后 离心分离。然后取上层清液,用紫外及可见分光光度计测定黄药残余浓度。
2 结果与讨论
2.1 前驱体的热重-差热分析
在制备前驱体的过程中,用0.10 mol·L-1硝酸铜溶液作为铜源,用25%氨水做配位剂,用0.3 mol·L-1NaOH 溶液作沉淀剂,制备得到氢氧化铜沉淀,经80 ℃干燥后可视为氧化铜前躯体。前驱体的TGDTA 测定分析结果如图1 所示:
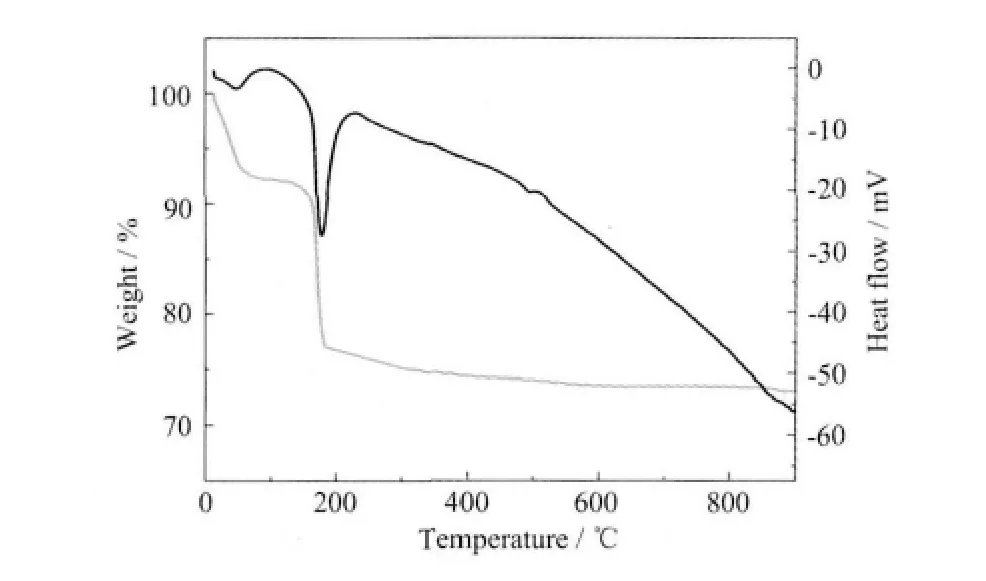
图1 制备纳米CuO 前驱体的TG-DTA 分析曲线Fig.1 TG-DTA curves for the precursor of nanoscale copper oxide
由前驱体的热重-差热分析曲线可以看出,前驱体在20~90 ℃范围内有失重现象,并且相对应的差热曲线有明显的吸热峰,证明在这个温度区间内存在前驱物中部分游离水和氨的脱离。此后随温度升高,热重曲线在较窄的温度区间出现平台,对应这个温度区间内前驱物几乎没有质量损失。在大约100 ℃以后又出现前驱物的失重以及相应的吸热曲线,由于此次吸热峰强度明显大于第一次吸热峰的强度,可以判断这个区间内的吸热较前一次吸热大得多,说明这次吸热伴随着更强的分子间作用力的断开,从热重曲线可以看出此区间失重率为18.20%说明前驱物在这个阶段很可能发生了如下反应:

反应结束后便得到CuO 粉体(理论失重率为18.45%)。在热重曲线中显示,大约在370~400 ℃时完成由前驱物到目标产物的转变。
2.2 纳米CuO 的XRD 分析及粒径控制研究
我们要利用醇-氨水体系制备纳米CuO 粉体,并利用不同的醇控制产物的粒径。在不同醇-氨水体系中制备的纳米CuO 的XRD 衍射图谱如图2 所示。图2 中a、b、c 依次是乙醇-氨水体系、正丁醇-氨水体系和正辛醇-氨水体系得到的纳米CuO 的XRD 谱图。从图中可以看出,每一种方法制备的CuO 都 与标准CuO 的XRD 衍 射图样(PDF No.89-5898)一致,为结晶良好的纯相CuO,表明在不同的溶液体系中,均可以制备较纯净的纳米CuO。而且随醇-氨水体系中醇的链长的增加,图谱中特征衍射峰的峰宽依次变宽,辛醇-氨水体系制备的CuO的峰最宽,由Scherrer 公式可知,这种体系制备的纳米CuO 的粒径最小。

图2 不同醇-氨水体系制备出的纳米CuO 的XRD 图Fig.2 XRD patterns of nano copper oxide prepared in different alcohol -ammonia system

表1 在不同溶剂体系所制备纳米CuO 的比表面积Table 1 Surface area of copper oxide nano particles prepared in different solvent system
我们将不同醇-氨水体系制备的纳米CuO 粉体进行BET 分析,测得样品的比表面积如表1 所示;由公式: d=6/(ρ×SBET)计算得到各物质的粒径,其中式中d 为颗粒直径,ρ 为固体颗粒的密度,SBET为固体的比表面积。
从上表中可以看出,所制得的产物在不同的溶剂体系呈现出比较强的规律性: 在水溶液中和在聚乙二醇(PEG)水溶液中生成的产物粒度较大;而在醇-氨水溶剂中,乙醇-氨水作为溶剂制得的产物比表面积最小,粒度最大;正丁醇-氨水作为溶剂制得的产物比表面有所增加,粒度相比乙醇-水作为体系制备的产物减小; 正辛醇-氨水作为溶剂制得的产物比表面积最大,粒度最小。
氧化铜颗粒直接受其前躯体颗粒大小影响,铜离子在水溶液中极易发生水解而生成氢氧化铜沉淀,这种沉淀颗粒的迅速生成和聚集往往会生成亚微米甚至微米数量级的沉淀聚集颗粒。欲制备纳米颗粒,就必须设法阻止沉淀颗粒的团聚。通过添加聚乙二醇可以起到分散沉淀颗粒防止沉淀聚集的作用。由于在水溶液中氨分子易与铜离子生成铜氨络离子,在溶液中氢氧根离子逐渐增加过程中,生成氢氧化铜沉淀和铜氨络离子之间的竞争反应可以有效地控制铜离子的释放速度,从而避免铜离子的迅速水解。因此,氨分子的存在应有益于生成较小的氢氧化铜沉淀颗粒。图3 是通过软件Medusa 模拟的铜离子-氨水体系的溶液组分随pH 值的变化示意图,相关反应平衡常数来自Medusa 软件数据库[31]。在试验条件下(铜离子和氨分子浓度分别为44 mmol·L-1和710 mmol·L-1),相关的溶液化学反应和有关平衡常数在表2 列出。
从图3 可以看出,当pH 小于10.6 时,溶液中Cu(NH3)42+是优势组分; 而pH 介于10.6~11.5 之间时,Cu(NH3)3OH+是优势组分;当pH 大于11.5 后Cu(OH)2(s)成为优势组分。在强碱性pH,由于溶液中CuNH3(OH)3-的生成,Cu(OH)2(s)沉淀会发生溶解。在氨水中,通过添加氢氧化钠,氢氧化铜沉淀的生成反应如图4 所示。
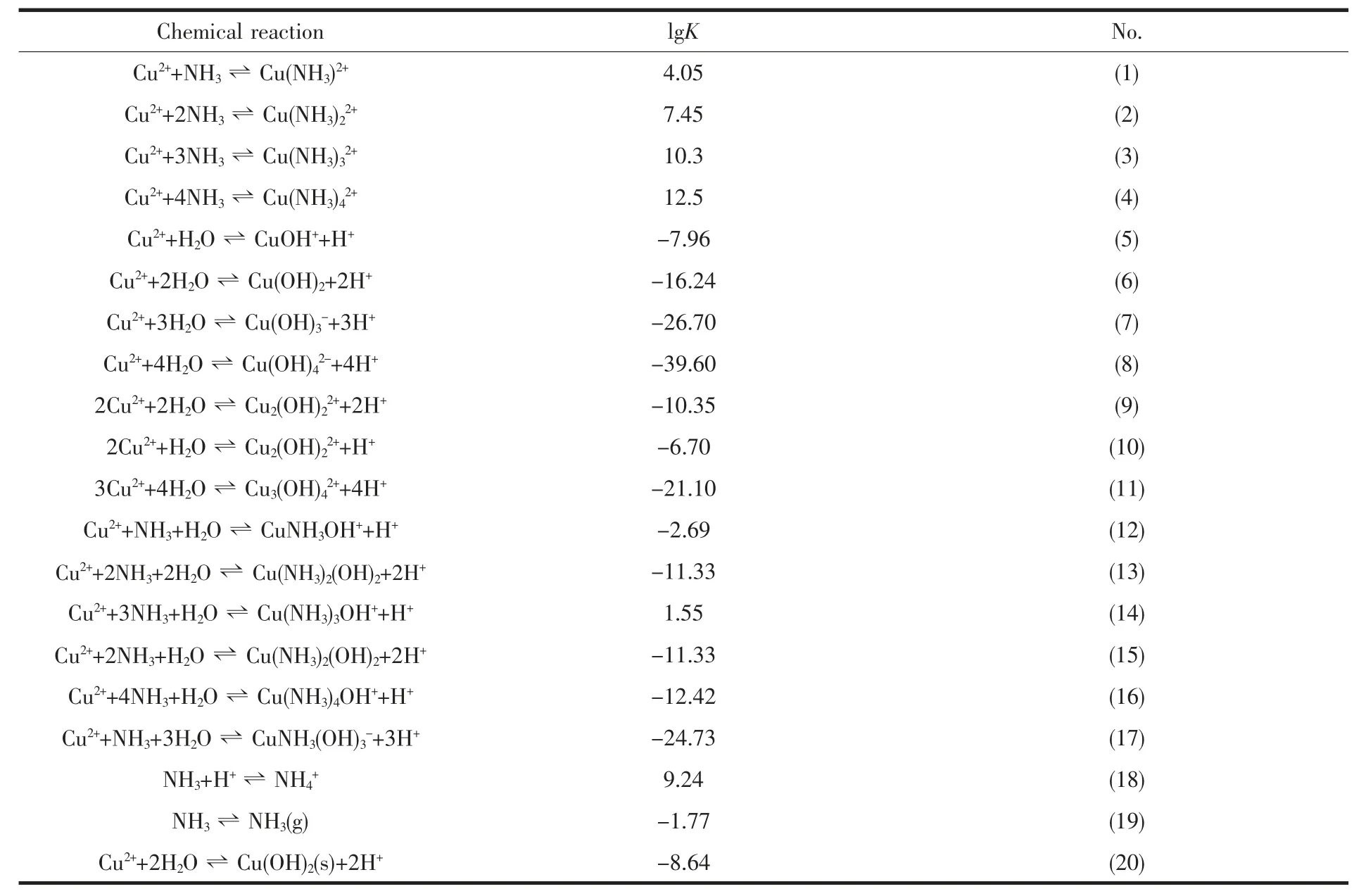
表2 在Cu2+-NH3-H2O 体系中的溶液化学反应和相应的平衡常数Table 2 Solution chemical reactions in Cu2+-NH3-H2O system and corresponding equilibrium constant
从图4 可看出,随着添加的氢氧根离子浓度的增加,溶液中含铜优势组分逐渐由Cu(NH3)42+转换成Cu(NH3)3OH+。当添加的氢氧根离子浓度为20 mmol·L-1时,氢氧化铜沉淀生成。随着添加的氢氧根离子浓度的进一步增加,氢氧化铜沉淀变成优势组分。当添加的氢氧根离子浓度高于130 mmol·L-1后,氢氧化铜沉淀含量达到最高值。由于氢氧根离子和氨分子之间对于铜离子的竞争反应,可能导致生成粒度较小的氢氧化铜沉淀颗粒。
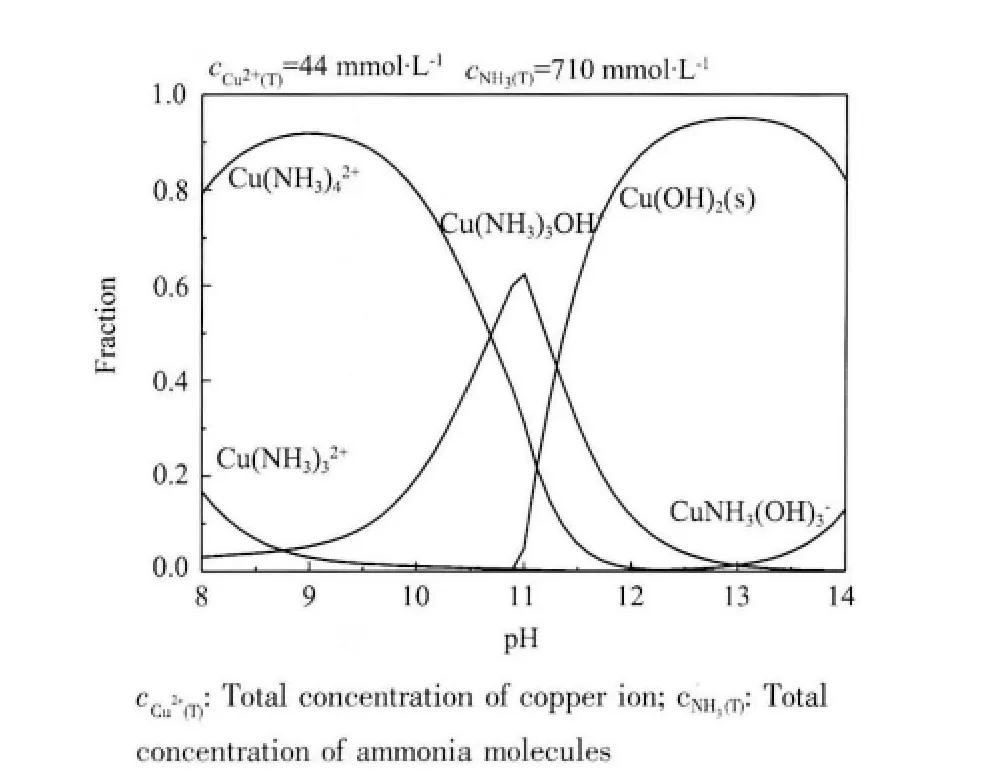
图3 铜离子在氨水中的组分分布与溶液pH 的关系Fig.3 Species distribution in Cu2+-NH3-H2O system as a function of pH

图4 氢氧化铜沉淀的生成与氢氧根离子添加量的关系Fig.4 Species distribution in Cu2+-NH3-H2O system as a function of cOH- addition
根据实验数据和实验结果我们可以推断,在前驱体成核的过程中,溶剂中的醇分子链可以有效的阻止晶核之间的团聚,这种阻碍作用随着醇的链长的增长而加剧,而且醇水介质中的传质比纯水中慢,从而得到分散较好,粒径较小的前驱体。前驱物经煅烧后得到呈现粒径规律性变化的纳米CuO 粉体。
2.3 纳米CuO 表面硫化前后样品的表征分析结果
在200 ℃下将纳米CuO 与单质硫混合研磨均匀煅烧90 min,得到黑色粉末为表面硫化的氧化铜CuO/CuS 样品,其XRD 衍射测定结果如图5(b)所示。从图5(b)可以看出,所有的衍射峰均与图5(a)所示的CuO 的XRD 标准谱图一致,表明硫化反应只发生在CuO 表面,由于并未发生体相硫化,故硫化产物的XRD 衍射峰呈现CuO 的图样。而从红外光谱的测定结果(图6)可以看出确实有硫化铜在氧化铜表面生成。由此可见,硫化产生的CuS 不足以使产物产生X-射线衍射的变化,可以推测硫化只是发生在CuO 的表面。利用这种方法可以对CuO 进行表面硫化。

图5 CuO 在200 ℃硫化90 min 前后的XRD 图Fig.5 XRD patterns of CuO before and after 90 min surface sulphidization at 200 ℃
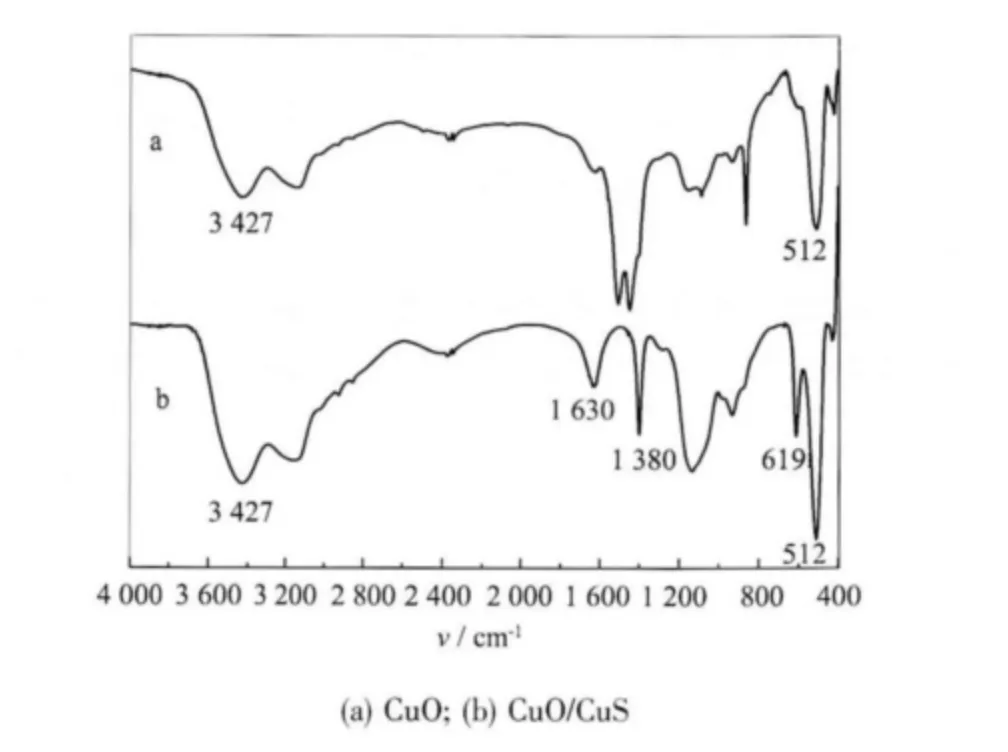
图6 CuO 硫化前后的红外光谱Fig.6 FT-IR spectrum of CuO and surface sulphidization CuO
纳米CuO 表面硫化前后的红外光谱对比如图6 所示,图6(a)为CuO 的红外光谱,图6(b)为CuO/CuS 的红外光谱。谱图中3 427 和1 627 cm-1处吸收峰为样品中的吸附水的伸缩振动和弯曲振动产生,在1 378 cm-1处的吸收峰由结晶水的弯曲振动产生的。表面硫化前后的红外谱图在指纹区出现了明显的差别,图6(a)中,512 cm-1处为CuO 的Cu-O伸缩振动产生;图6(b)中,除了CuO 的Cu-O 伸缩振动外,在619 cm-1处出现CuS 的Cu-S 伸缩振动峰,表明在CuO 硫化后,样品中同时存在CuO 和CuS,进一步证明利用上述方法可以对CuO 成功进行表面硫化,得到表面包覆CuS 的CuO。

图7 200 ℃硫化150 min CuO/CuS 的XRD 图Fig.7 XRD pattern of nano copper oxide after sufidizing 150 min at 200 ℃
若延长反应时间,在200 ℃下将纳米CuO 与单质硫混合研磨均匀煅烧150 min,得到硫化产物的XRD 衍射谱图如图7 所示,其衍射花样与纯氧化铜的有明显的不同。与标准卡片对比可知,硫化产物为多硫化铜的混合物,其中主要是CuS 和Cu9S5(铜与硫比例接近2∶1)产生的衍射峰,表明CuO 晶面在渐渐消失,晶相发生变化,发生体相硫化。
由于硫源是单质硫,氧化铜的硫化过程可能是通过硫的歧化反应实现的,部分硫被还原成-2 价,部分硫被氧化成+4 价,如下式所示:
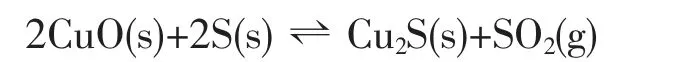
反应生成的最终稳定产物可能是硫化亚铜和二氧化硫,而CuS 则可能为不同反应动力学阶段的过渡产物。
综合上述实验我们可以得出,在其他反应条件相同的情况下,单纯通过调节反应时间就可以有效的控制硫化的效果,随着反应时间的延长,表面硫化逐渐转变为体相硫化。这说明硫化是硫化铜在表面生成并向氧化铜体相扩散的过程,而在适当的温度条件下,氧化铜的硫化程度完全取决于硫化反应时间。
2.4 CuO 表面硫化前后对乙基黄原酸钾的吸附分析
按照实验部分讲述的方法和步骤,将CuO 和CuO/CuS 分别与乙基黄原酸钾吸附后,溶液中剩余乙基黄原酸钾的紫外吸收光谱如图8 所示,其中图8(a)为吸附前乙基黄原酸钾溶液的吸收光谱,图8(b)为CuO 吸附乙基黄原酸钾后溶液的吸收光谱,图8(c) 为CuO/CuS 吸附乙基黄原酸钾后溶液的吸收光谱。

图8 吸附后乙基黄原酸钾的紫外吸收光谱Fig.8 UV spectra of residual potassium ethyl xanthate
从图中可以看出,与吸附前相比,剩余乙基黄原酸钾的浓度明显变小,表明CuO 和CuO/CuS 都对乙基黄原酸钾有较强的吸附能力。对比(b)和(c)可以发现,表面硫化可以增强CuO 对乙基黄原酸钾的吸附能力,这也是选矿工艺中通过表面硫化来提高CuO浮选效率的理论依据。
3 结 论
在醇-氨水体系中利用铜离子的配位沉淀法可以制备出粒度可控、 结晶态较好的纳米CuO 粉体,其粒度随着醇链长的增加而减小。
在氮气保护下加热单质硫与纳米CuO 的混合物,可以成功对纳米CuO 进行表面硫化。XRD 和红外光谱分析表明,硫化是硫化铜在氧化铜表面生成并向体相扩散的过程。在适当的温度条件下,通过调节反应时间可以有效地控制硫化程度, 使氧化铜逐渐从表面硫化向体相硫化转变。
[1] Li X P, Wang Y, Lei Y, et al. RSC Advances, 2012,2(6):2302-2307
[2] Yang M, He J, Hu X, et al. Environ. Sci. Technol., 2011,45(14):6088-6094
[3] Zhang F, Zhu A W, Luo Y P, et al. J. Phys. Chem. C,2010,114(45):19214-19219
[4] Hoa N D, Quy N V, Tuan M A, et al. Physica. E, 2009,42(2):146-149
[5] Li J, Y, Xiong S L, Xi B J, et al. Cryst. Growth Des., 2009,9(9):4108-4115
[6] Chen J, Wang K, Hartman L, et al. J. Phys. Chem. C, 2008,112(41):16017-16021
[7]Zhang J,Liu J,Peng Q,et al.Chem.Mater.,2006,18(4):867-871
[8] WANG Ling(王岭), HAO Zeng-Chuan(郝增川), DAI Lei(戴磊),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28(1):81-87
[9] Xu L, Sithambaram S, Zhang Y, Chen C H, et al. Chem.Mater., 2009,21(7):1253-1259
[10]Chen C, Qu J, Cao C, et al. J. Mater. Chem., 2011,21(15):5774-5779
[11]Zhou M, Gao Y, Wang B, et al. Eur. J. Inorg. Chem.,2010,2010(5):729-734
[12]YANG Xiao-Yan(杨晓艳), SUN Song(孙松), DING Jian-Jun(丁 建 军), et al. Acta. Phys. -Chim. Sin.(Wuli Huaxue Xuebao), 2012,28(8):1957-1963
[13]GUO Xiao-Ming(郭晓明), MAO Dong-Sen(毛东森), LU Guan-Zong(卢 冠 忠), et al. Acta Phys. -Chim. Sin.(Wuli Huaxue Xuebao), 2012,28(1):170-176
[14]HENG Qiu-Li(衡秋丽), XIAO Feng(肖峰), LUO Jian-Min(骆 建 敏), et al. Chinese J. Inorg. Chem. (Wuji Huaxue Xuebao), 2009,25(2):359-363
[15]SHAO Qian (邵谦), WANG Xiao-Jie(王小杰), GE Sheng-Song(葛圣松),et al.Chinese J. Inorg. Chem. (Wuji Huaxue Xuebao), 2012,28(5):1043-1049
[16]Zhang X J, Wang G F, Liu X W, et al. J. Phys. Chem. C,2008,112(43):16845-16849
[17]Miao X M, Yuan R, Chai Y Q, et al. J. Electroanal. Chem.,2008,612(2):157-163
[18]Jia W, Guo M, Zheng Z, et al. Electroanal., 2008,20(19):2153-2157
[19]Qiu G H, Dharmarathna S, Zhang Y S, et al. J. Phys. Chem.C, 2012,116(1):468-477
[20]Li J Y, Xiong S L, Pan J, et al. J. Phys. Chem. C, 2010,114(21):9645-9650
[21]YIN Yi-Dong(尹贻东), HOU Hai-Ge(侯海鸽), FAN Nai-Ying(范乃英), et al. Chinese J. Inorg. Chem. (Wuji Huaxue Xuebao), 2010,26(2):293-299
[22]Chen J, Wang K, Hartman L, et al. Phys. Chem. C, 2008,112(41):16017-16021
[23]Xiang J Y, Tu J P, Zhang L, et al. J. Power Sources,2010,195(1):313-319
[24]Li D, He Y J, Wang S. J. Phys. Chem. C, 2009,113(30):12927-12929
[25]Li T, Ai X P, Yang H X. J. Phys. Chem. C, 2011,115(13):6167-6174
[26]Zhang W X, Li M, Wang Q, et al. Adv. Funct. Mater., 2011,21(18):3516-3523
[27]Wang G L, Huang J C, Chen S L, et al. J. Power Sources,2011,196(13):5756-5760
[28]Liu B, Zeng H C. J. Am. Chem. Soc., 2004,126(26):8124-8125
[29]ZHU Jun-Wu(朱俊武), ZHANG Wei-Guang(张维光),WANG Heng-Zhi(王 恒 志), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2004,20(7):863-867
[30]WANG Wen-Liang(王文亮), LI Dong-Sheng(李东升),WANG Zhen-Jun(王 振 军), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2002,18(8):823-826
[31]Puigdomenech I. MEDUSA ver 2.0. Stockholm, Sweden:Royal Institute of Technology, 1999.
