铈锆固溶体的表面碱性对催化剂中Pd 烧结性能的影响
2013-08-20肖益鸿夏笑莹阳艳玲蔡国辉魏可镁
肖益鸿 夏笑莹 阳艳玲 蔡国辉 郑 勇 魏可镁
(福州大学化肥催化剂国家工程研究中心,福州 350002)
贵金属钯是净化催化剂中最常用的活性组分[1-3],通常将其制备成负载型催化剂。由于净化催化剂通常是直接装在发动机歧管后以达到快速起燃,因此催化剂所处的温度高达1 000 ℃[4]。在这样工作条件下,使得贵金属活性组分在表面聚集烧结,催化剂活性位减少因而活性降低[5]。因此,如何抑制高温下活性组分的烧结行为一直是汽车尾气催化净化器性能需要解决的一个重要问题。
负载型催化剂的稳定性与活性组分和载体间的相互作用有关。Baker 等[6]认为贵金属与载体间相互作用太弱易发生贵金属的烧结,他们在对Pd/Al2O3和Pd/TiO2的金属载体相互作用研究中,发现Al2O3载体与Pd 的相互作用相对较弱,Pd 在Al2O3比其在TiO2上更容易发生烧结。
CeO2其不仅具有储氧能力,还能促进贵金属粒子的分散稳定作用[7],其常作为汽车尾气净化催化剂的载体。文献报道了Pd/CeO2催化剂中金属载体间通过相互作用形成了Pd-O-Ce 键[8-9],这种作用可以将Pd 粒子锚定在载体表面,从而可抑制Pd 粒子的高温烧结。Nagai 等[10]在对负载型Pt 催化剂研究中,也发现金属与载体形成的Pt-O-Ce 键的强度随着氧电子密度的增大而增大。他们通过对Pt/CZY(铈锆钇复合氧化物)催化剂研究,认为通过对Pt 的锚定作用,可使其保持高氧化态,从而抑制了Pt 粒子的烧结。而Hidetoshi 等[11]研究认为氧化物的碱性和暴露在表面的O2-离子相关。
铈锆固溶体不仅具有比CeO2更好的储氧能力和热稳定性[12-13],我们认为其表面碱性也将会影响金属载体相互作用。因此,本工作合成出系列CexZr1-xO2(0≤x≤1)固溶体,以其为载体制备负载Pd型催化剂。利用原位红外漫反射研究了CO2在载体上吸附脱附行为,考察载体碱性变化对Pd/CexZr1-xO2催化剂中Pd 的分散度以及热稳定性的影响。
1 实验部分
1.1 样品的制备
以(NH4)2Ce(NO3)6、Zr(NO3)4·5H2O 为原料,按nCe/nZr为0,0.25,0.50,0.67,0.75 和1 配成溶液,再加入3 倍化学计量比的尿素,搅拌均匀后置于反应釜中,180 ℃反应60 h。冷却后离心分离、洗涤,110 ℃烘干4 h,900 ℃焙烧8 h,即得到CexZr1-xO2样品(系列代号为C), 当x 为0,0.25,0.50,0.67,0.75 和1 时对应的样品编号为C0,C25,C50,C67,C75 及C100。
上述595~177 μm(30~80 目)的CexZr1-xO2样品为载体,以Pd(NO3)2为前驱体,采用浸渍法制备Pd/C 催化剂,Pd 的负载量均为1.83at%(Pd 含量采用原子吸收光谱分析测定)。催化剂经110 ℃干燥后于600 ℃焙烧4 h,得到催化剂简记为C-F,相应的催化 剂 编 号 为C0-F,C25-F,C50-F,C67-F,C75-F 及C100-F;经850 ℃焙烧4 h,得到催化剂简记为C-A,相 应 编 号 为C0-A,C25-A,C50-A,C67-A,C75-A 及C100-A。
1.2 样品的表征
样品粉末X 射线衍射分析在荷兰PANalytical公司Xpert Pro X 射线粉末衍射仪上进行,采用XCelerator 探测器,Co Kα(λ=0.117 902 nm) 为辐射源,管压40 kV,管流20 mA,扫描步长0.033 3°,扫描速率为40 s·step-1。
BET 比表面积测试在Micromeritics Tristar 3000型快速比表面积与孔隙率分析仪上进行, 在液氮温度(77 K)下进行N2物理吸附,样品测定前经300℃抽真空预处理3 h。
CO 脉冲化学吸附和CO2程序升温脱附(CO2-TPD)在美国Micromeritics 公司Auto chemⅡ2920 分析仪上进行,TCD 为检测器。CO 脉冲化学吸附是将试样在500 ℃下用H2预还原30 min,然后在He 气氛中吹扫至室温,吸附气组成为体积分数5%CO/He的混合气,高纯He 为载气,采用脉冲进样至峰面积相同, 然后根据累积的气体吸附量按计量比nCo/nPd=1∶1 计算得到表面Pd 原子数,表面Pd 原子数乘以Pd 原子的横截面积得Pd 的表面积;表面Pd 原子数除以催化剂中总Pd 原子数得Pd 的分散度[14]。CO2-TPD 是将试样于He 气氛中300 ℃处理30 min 后降至 100 ℃,以高纯CO2脉冲吸附至饱和,He 吹扫30 min 待基线稳定后,以He 为载气进行程序升温脱附,升温速率10 ℃·min-1,程序升温范围:100~600 ℃。
原位漫反射红外光谱分析(in situ-DRIFTS)在美国Thermo 公司的Nicolet 6700 型红外光谱仪上进行,使用MCT 检测器,分辨率为2 cm-1,扫描次数为60,波数范围1 200~1 800 cm-1。取粉末样约0.2 g,装填至样品池中,压平表面,在N2气氛中(25 mL·min-1)于300 ℃处理1 h 并降至100 ℃,采集背景,通 高 纯CO2(25 mL·min-1) 吸 附30 min,N2吹 扫30 min,在N2气氛中(25 mL·min-1)以约10 ℃·min-1的速率升温至600 ℃,每隔20 ℃采集一张谱图。
2 结果与讨论
2.1 C 系列样品的物相分析
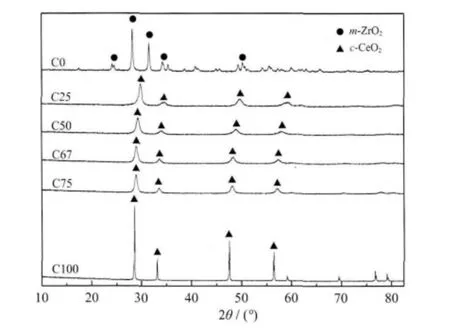
图1 C 系列样品的XRD 图Fig.1 XRD patterns of C series samples
图1 为C 系列样品的XRD 图。C0 样品在24.1°、28.2°、31.5°、34.2°、50.1°等处出现了单斜相斜锆石(m-ZrO2)的特征峰(PDF 卡片号00-024-1165),说明 其 为 单 斜 相 斜 锆 石;C25~C100 样 品 在28.6°、33.1°、47.5°、56.3°等处出现了立方相方铈矿(c-CeO2)的特征峰(PDF 卡片号00-043-1002),而且其特征衍射峰随着锆含量的增加向高角度方向移动,这主要归因于Zr4+进入到CeO2的晶格中,由于Zr4+的离子半径(0.084 nm)小于Ce4+的离子半径(0.097 nm),造成晶格收缩,晶胞参数变小。因此,铈锆固溶体随Zr4+增多衍射角增大。上述结果表明所合成的样品C0、C25、C50、C67、C75 及C100 均为铈锆固溶体。
2.2 Pd/C 样品的分散度
从表1 可看出以所合成的固溶体制备的钯催化剂,其分散度随着铈含量的增加逐渐增大,Pd 粒径随着铈含量的增加而减小,相应的Pd 活性表面积随着铈含量的增加而增大,其中C75-F 样品钯分散度较高为14.0%,样品具有较小的粒径8.0 nm 和较大的Pd 表面积62.2 m2·g-1。但C 系列样品的比表面积不随铈含量的增加而增大,且C100-F 的低分散度仅为2.0%,Pd 的分散度显著降低了,观察C100 样品的X-射线衍射峰非常尖锐,可能原因是以过高铈含量制备的催化剂其晶粒长大,这可能与载体的比表面积以及载体物相组成有关。C-A 系列催化剂中Pd 的分散度均低于C-F 系列催化剂的分散度。催化剂经850 ℃焙烧后其活性组分Pd 表面积与新鲜催化剂相比均有不同程度的降低,且活性组分表面积降低程度随着铈含量的增加而减少。同时可以看出,以C75 为载体的制备的催化剂Pd 粒子的高温烧结程度相对较小。
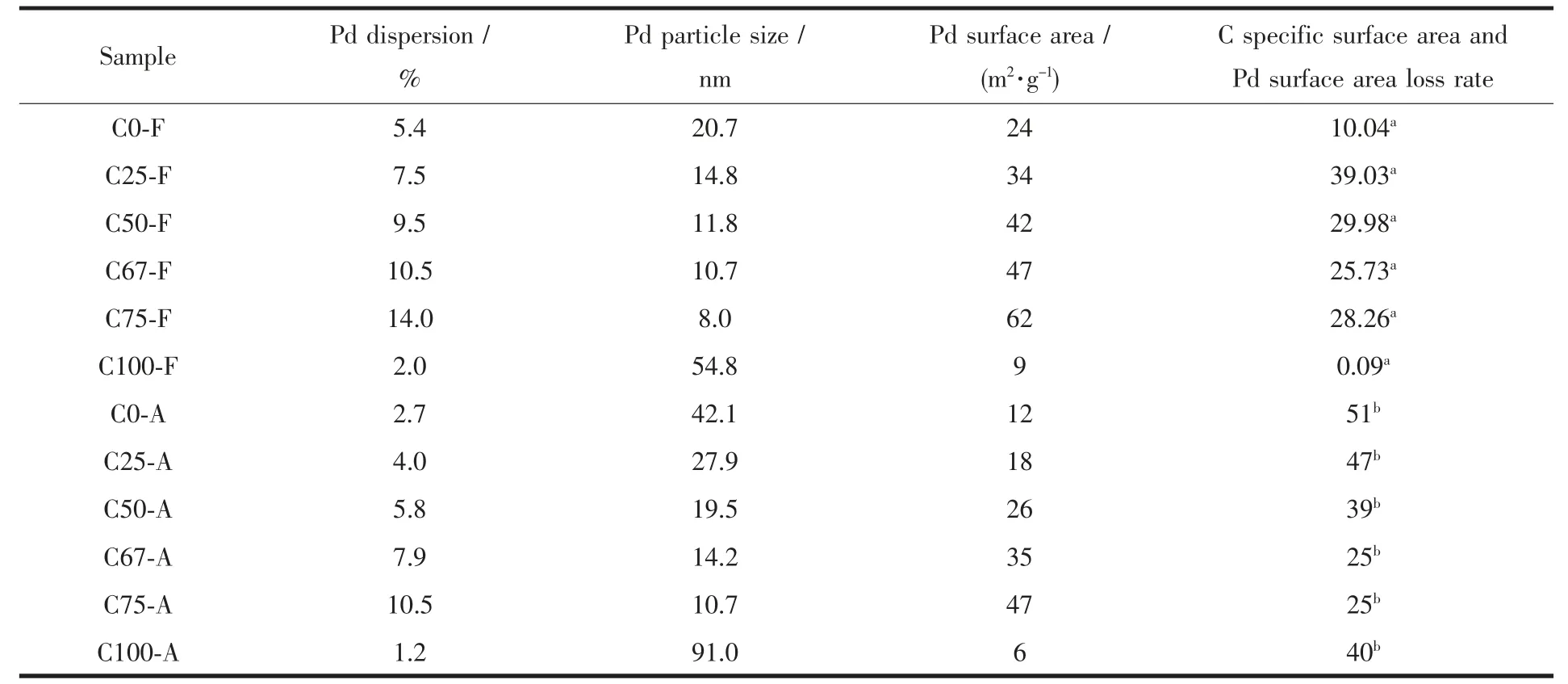
表1 C-F 及C-A 系列样品的Pd 分散度数据Table 1 Pd dispersion of C-F and C-A series samples
2.3 原位红外光谱
氧化物的表面羟基、氧离子会与CO2形成碳酸氢盐或碳酸盐物种,所以氧化物的碱性可以用CO2作为探针分子进行研究[15]。应用原位红外漫反射程序升温脱附(in situ DRIFTS-TPD)实验技术,可以实时跟踪CO2的脱附行为,进而可以分析氧化物表面碱性位的特性。CO2吸附在碱性-OH 基团上会产生表面碳酸氢盐,吸附在金属离子和邻近的氧离子上形成双齿碳酸盐,吸附在金属离子上并与过量的氧生成单齿碳酸盐[16-17]。图2 为C 系列样品经N2吹扫后吸附CO2测得的红外光谱,相应吸附物种的结构示意式及谱峰如表2 所示。
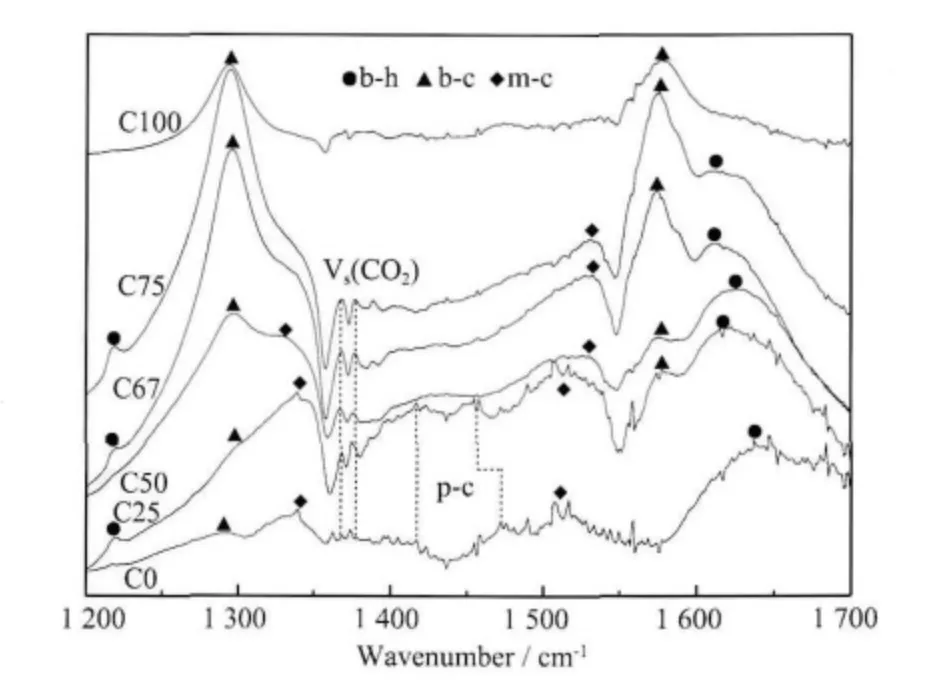
图2 C 系列样品的原位红外光谱图Fig.2 In situ DRIFTS spectra of C series samples
表2 中C0 和C25 样品在红外谱图中呈现双齿碳酸氢盐、双齿碳酸盐、单齿碳酸盐的吸收带外,还存在多齿碳酸盐p-c 吸收带,C0 的峰位于1 419 和1 475 cm-1;C25 的在1 419 和1 457 cm-1出现吸收带,说明这两个样品表面与其他样品比较具有不同的碱性位。C100 样品其吸附CO2后仅形成双齿碳酸盐物种,可能是其比表面较低的缘故。另外,C0~C75样品1 368 和1 376 cm-1波数的2 个强度较弱但是峰形尖锐的峰是CO2的对称伸缩模式,在图2 中用Vs(CO2)表示[19]。
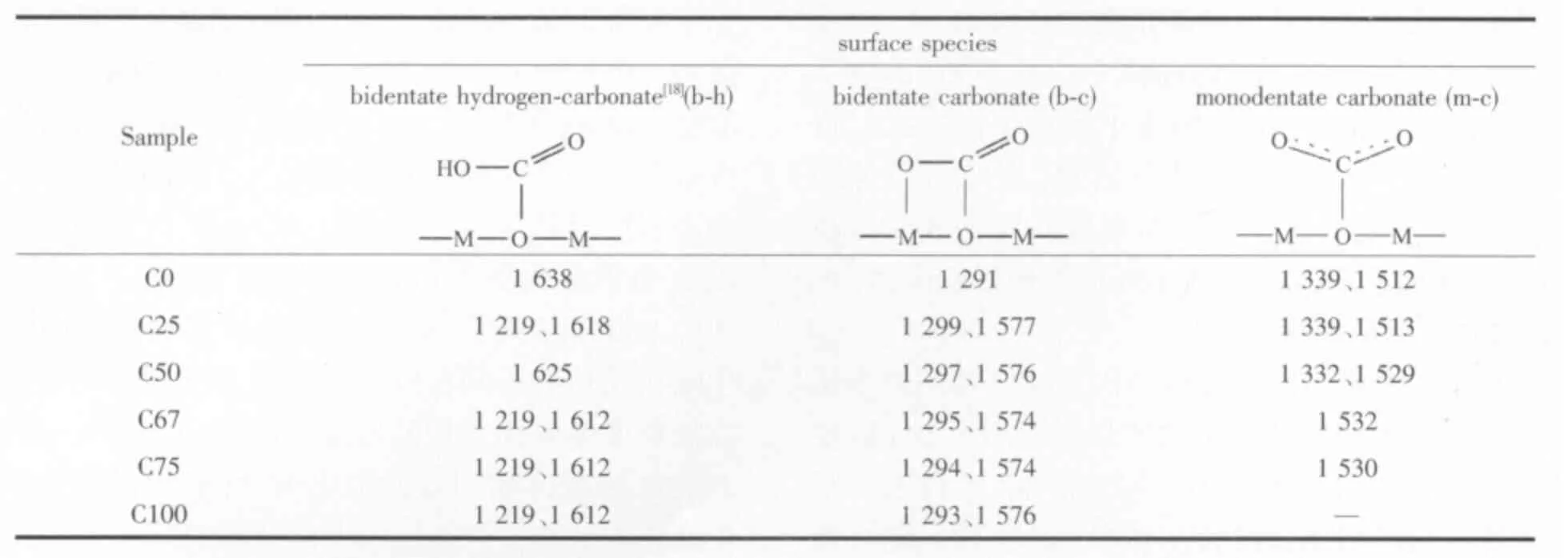
表2 C 系列样品表面吸附物种的谱峰(cm-1)Table 2 IR bands of surface species on C series samples(cm-1)
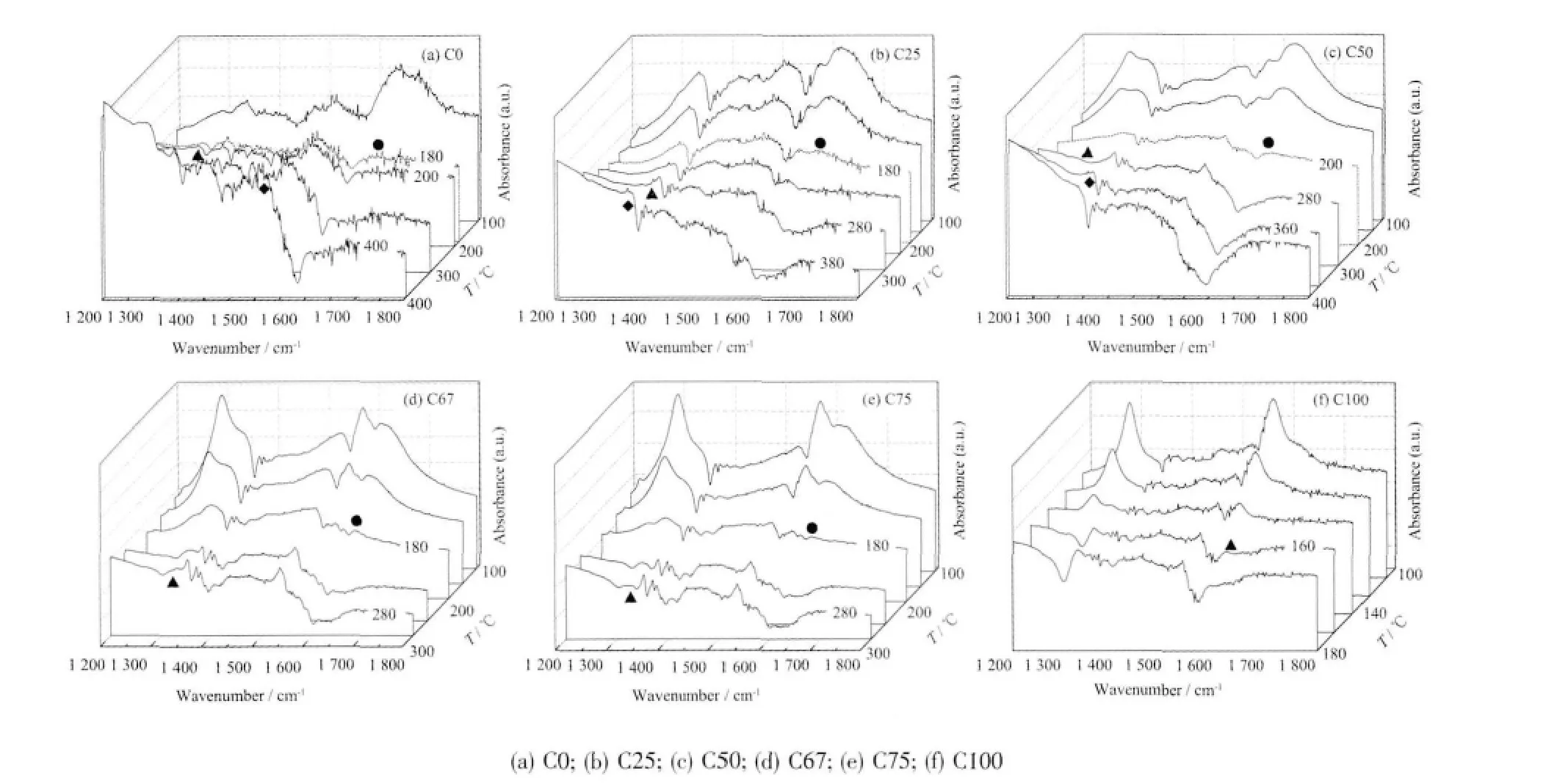
图3 C 系列样品的in situ DRIFTS 图谱Fig.3 In situ DRIFTS spectra of C series samples
图3 为C 系列样品吸附CO2后的in situ DRIFTS-程序升温脱附图,其中双齿碳酸氢盐b-h、双齿碳酸盐b-c、单齿碳酸盐m-c 各吸附物种相应的特征峰波数依次用圆形(●)、三角形(▲)、菱形(◆)标出。从图中可以看出,随着温度的升高,各物种的特征吸附峰逐渐变小,直至消失。从图中标有温度的曲线可以看出各吸附物种特征吸附峰消失的温度,双齿碳酸氢盐在200 ℃以内,双齿碳酸盐在300 ℃以内,单齿碳酸盐在400 ℃以内。这说明样品表面吸附CO2所形成的各物种随着温度的升高而不断脱附,而物种特征峰消失的温度我们可以认为是其分解温度点。因此由图3 可以得到各样品表面吸附物种bh、b-c、m-c 分解温度,如表3 所示。根据b-h、b-c、m-c吸附物种的完全分解温度特性,可以通过以下的CO2-TPD 表征来大致判断各吸附物种的量。
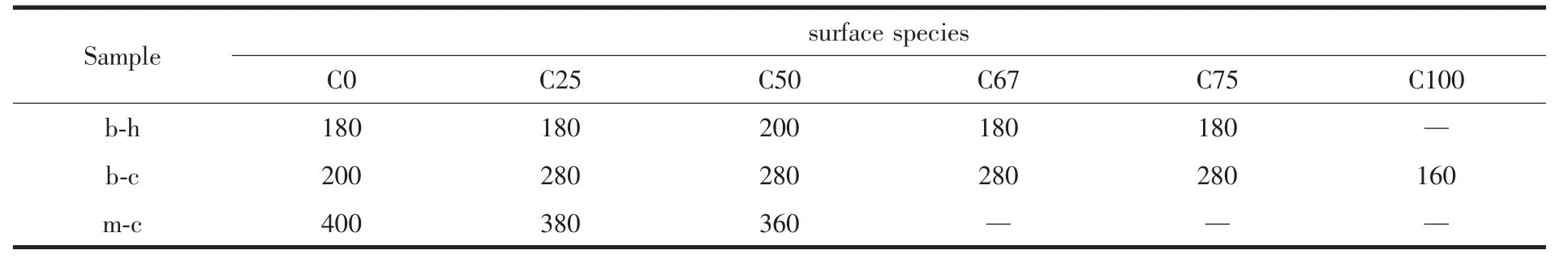
表3 C 系列样品b-h、b-c、m-c 的完全分解温度(℃)Table 3 Complete decomposition temperature of b-h、b-c、m-c on C series samples(℃)
2.4 CO2-TPD 表征
图4 是C 系 列 样 品 的CO2-TPD 图,C0 有3 个脱附峰,峰温依次是158、208、399 ℃;C25 有3 个脱附峰,峰温依次是170、275、376 ℃;C50 样品3 个脱附峰温依次是188、271、365 ℃,但其后2 个峰面积与C0、C25 相比显著降低;C67 样品只有两个脱附峰,峰温依次是185、274 ℃;C75 样品的峰温依次是180、266 ℃, C100 样品的峰温是148 ℃,峰面积小。根据上述in situ-DRIFTS 的表征结果,我们将200℃以下的CO2脱附峰归属于双齿碳酸氢盐(b-h)的分解,200~300 ℃的CO2脱附峰归属于双齿碳酸盐(bc)的分解,300 ℃以上的峰归属于单齿碳酸盐(m-c)的分解。从峰面积上看,纯铈样品C100 因为其晶粒大、比表面积小,所以吸附的CO2量小故脱附峰面积小。在铈锆载体吸附CO2形成的拟碳酸盐中,随着铈含量的增加,单齿碳酸盐和双齿碳酸盐逐渐减少,而双齿碳酸氢盐逐渐增多,其中C75 的以双齿碳酸氢盐吸附的CO2量较高,说明其具有较多的弱碱性位。
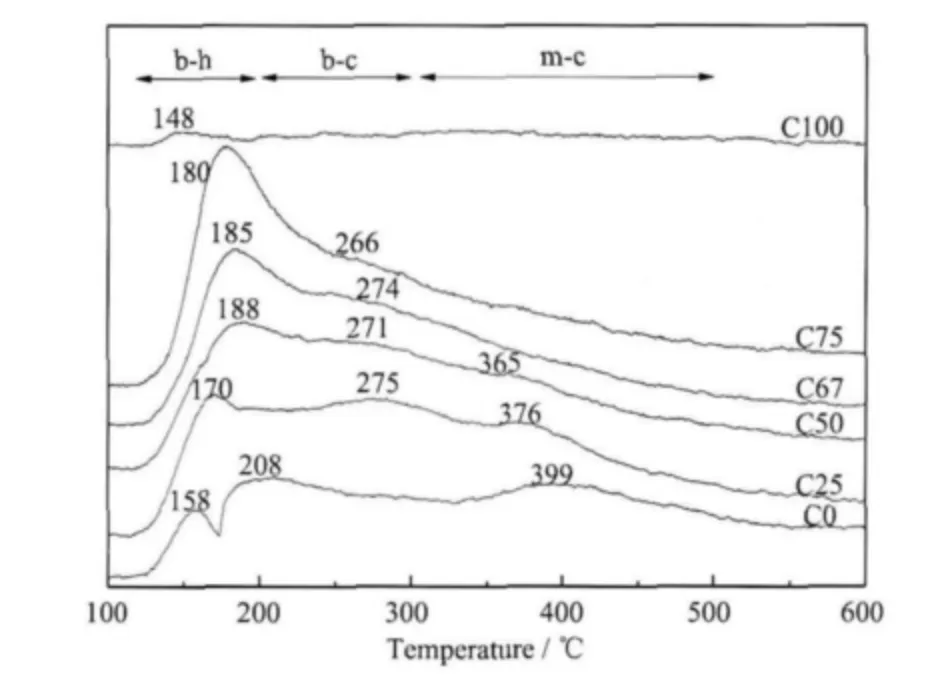
图4 C 系列样品的CO2-TPD 图Fig.4 CO2-TPD profiles of C series samples
由于CO2在载体表面的弱碱性位上会形成碳酸氢盐物种,根据上述分析说明C75 样品表面具有较多的弱碱性位。我们认为这种丰富的弱碱性位是抑制Pd 粒子高温烧结的关键因素。在铈锆固溶体中,Pd 粒子与CeO2之间有很强的相互作用,它们之间会形成Pd-O-Ce 键[20],通过锚定作用把Pd 粒子锚定在铈锆载体上,从而降低Pd 粒子的移动性,因而能达到抑制其高温烧结、提高金属分散度的作用。弱碱性位越多,表示载体表面O 的电子密度越大[21],形成的Pd-O-Ce 键相对也就越稳定,因此C75 样品具有相对较高的分散度以及较好的热稳定性。
3 结 论
以尿素为沉淀剂,采用均匀沉淀法合成出了CexZr1-xO2(0≤x≤1)铈锆固溶体,研究表明其弱碱性位能够有效抑制Pd 的烧结,载体弱碱性位的数量越多,其能更好地抑制Pd 的烧结行为。合成的Ce0.75Zr0.25O2载体具有较多的弱碱性位,以其为载体制 备 的Pd/Ce0.75Zr0.25O2催 化 剂,Pd 具 有 较 高 的 分 散度(14.0%),较显著地抑制了钯的烧结行为,具有较好的热稳定性。
[1] Shelef M, Mccabe R W. Catal. Today, 2000,62(1):35-50
[2] Muraki H, Zhang G. Catal. Today, 2000,63(2/3/4):337-345
[3] SHAO Qian(邵潜), LONG Jun(龙军), HE Zhen-Fu(贺振富),et al. Monolithic Catalysts and Monolithic Reactors(规 整 结构 催 化 剂 及 反 应 器). Beijing: Chemical Industry Press,2005:70
[4] Engler B, Koberstein E, Schubert P. Appl. Catal., 1989,48(1):71-92
[5] Miyoshi N, Matsumoto S, Ozawa M, et al. SAE(Society of Automotive Engineers),1989.DOI:10.4271/891970
[6] Baker R T K, Prestridge E B, Mcvicker G B. J. Catal., 1984,89(2):422-432
[7] Guo Y, Lu G, Zhang Z, et al. Catal. Today, 2007,126:296-302
[8] Satsuma A, Sato R, Osaki K, et al. Catal. Today, 2012,185:61-65
[9] Hosokawa S, Taniguchi M, Utani K, et al. Appl. Catal. A,2005,289:115-120
[10]Nagai Y, Hirabayashi T, Dohmae K, et al. J. Catal., 2006,242:103-109
[11]Nagamoto H, Shinoda E, Inoue H. Ind. Eng. Chem. Res.,1993,32:1790-1794
[12]GU Ying-Ying(古映莹), FENG Sheng-Sheng (冯圣生), LI Jin-Lin( 李 金 林), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2006,22(9):1623-1627
[13]ZHU Chang-Quan(朱昌权), LIANG Hong(梁红), LI Shu-Hua(李 树 华), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2011,27(6):1093-1100
[14]XIAO Yi-Hong(肖益鸿),LI Gui-Ping(李桂平),ZHENG Ying(郑瑛),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(1):61-66
[15]Lavalley J C. Catal. Today, 1996,27(3-4):377-401
[16]AurouxA,GervasimA.J.Phys.Chem.,1990,94(16):6371-6379
[17]Manchado M C, Guil J M,Mafia A P. Langmuir, 1994,10(3):685-691
[18]Pokrovski K, Kyeong T J, Alexis T B. Langmuir, 2001,17:4297-4303
[19]Marco D, Claude B, Jean C L. Phys. Chem. Chem. Phys.,1999,1:5717-5724
[20]Atsushi S, Ryota S, Kaoru O, et al. Catal. Today, 2012,185:61-65
[21]Mun S F, Ahmad Z A, Subhash B. Appl. Catal. B, 2010,100:365-377
