球孢白僵菌海藻糖-6-磷酸合成酶基因tps1真核表达载体的构建
2013-08-08翎1勃2
谢 翎1,2,黄 勃2
(1.安庆师范学院生命科学学院,安徽安庆 246011;2.安徽农业大学林学与园林学院,安徽合肥 230036)
酵母作为真核单细胞生物,兼有原核生物和真核生物的某些优点。与原核生物表达体系相比,利用酵母对外源基因进行真核表达具有诸多优势:繁殖迅速,培养廉价;酵母结构简单,便于进行异源基因的遗传操作;具有蛋白翻译后修饰能力[1];可在信使RNA翻译时去甲硫氨酸,避免药物使用中产生免疫反应。目前技术最成熟,应用最多的酵母表达系统是毕赤酵母(Pichia pastoris)。
pPIC9K质粒含有kan基因,使毕赤酵母具有G418抗性。G418抗性水平主要依赖整合的kan基因的数目。kan基因与表达盒间具有遗传连锁,因此从G418抗性可推断其所含有的插入基因拷贝数。
目前,国内外利用这种表达体系已成功表达多种异源蛋白[2-3]。但迄今为止,关于昆虫病原真菌海藻糖-6-磷酸合成酶基因tps1在毕赤酵母中实现真核表达的研究还未见报道。本研究首次构建了球孢白僵菌海藻糖-6-磷酸合成酶基因tps1真核表达载体,为海藻糖-6-磷酸合成酶的蛋白研究及大规模发酵生产奠定了基础。
1 材料和方法
1.1 材料
1.1.1 实验菌株与质粒
含球孢白僵菌海藻糖-6-磷酸合成酶基因tps1的质粒PMD18-T/Bbtps1,由实验前期构建。分泌型真核表达穿梭载体pPIC9K与大肠杆菌JM109菌株由本实验室保存。
1.1.2 试剂
TaqDNA聚合酶、PMD18-T载体、DNA胶回收试剂盒均购自大连宝生物公司。引物合成由上海生工生物工程有限公司完成。其他常用试剂均为国产分析纯试剂。
1.2 实验方法
1.2.1 Bbtps1基因的真核表达载体pPIC9K/tps1的构建
根据球孢白僵菌海藻糖-6-磷酸合成酶基因tps1序列及真核分泌型表达载体pPIC9K表达框及多克隆插入要求,设计上游引物TPS1BD1,下游引物TPS1BD2。通过PCR反应从质粒PMD18-T/Bbtps1(由前述实验构建)中扩增出带EcoRⅠ和NotⅠ酶切位点的Bbtps1 cDNA全长序列。
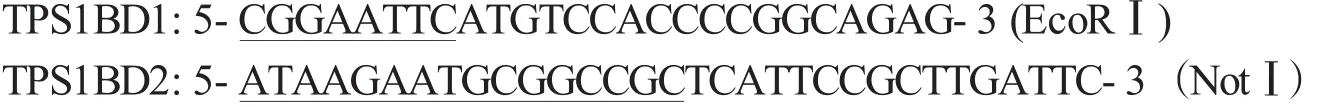
下划线部分序列分别带有限制性内切酶EcoRⅠ和NotⅠ的识别位点。
根据特异引物TPS1BD1、TPS1BD2对PMD18-T/Bbtps1质粒进行扩增。
酶切后的酵母表达质粒pPIC9K与目的基因片段用T4 DNA连接酶连接,连接产物转化宿主菌DH5α,转化菌液涂布在含卡那霉素的LB平板上过夜,挑选阳性克隆子用插入基因特异引物TPS1BD1、TPS1BD2进行PCR鉴定并送测序。
1.2.2 大肠杆菌感受态细胞的制备
制备方法参照《Molecular Cloning》所介绍的CaCl2法[4]。
1.2.3 DNA回收及克隆、测序
在紫外灯下切下含目的片段的凝胶块,用takara的胶回收试剂盒回收DNA。回收片段按试剂盒说明书连接到PMD18-T载体上。大肠杆菌感受态细胞制备及克隆转化具体方法参考《分子克隆实验指南》。阳性转化子委托上海英骏生物技术有限公司进行双向测序。
1.2.4 序列分析
序列分析用DNAMAN软件和NCBI网站(http://www.ncbi.nlm.nih.gov)上的BLAST程序完成。
2 结果与分析
在构建Bbtps1表达载体pPIC9K/tps1过程中,首先,扩增球孢白僵菌海藻糖-6-磷酸合成酶基因tps1 ORF的cDNA序列(1563bp,两边分别带EcoRⅠ和NotⅠ酶切位点)。将其双酶切后,通过T4连接与同样经过双酶切的质粒pPIC9K,构成质粒pPIC9K/tps1,转化DH5α。通过引物TPS1BD1、TPS1BD2对重组质粒的大肠杆菌转化子进行了PCR鉴定(图1),同时挑取PCR鉴定正确的转化子进行测序。序列分析结果表明,Bbtps1基因被正确地插入到pPIC9K质粒的表达框中。

图1 重组质粒pPIC9K/tps1 PCR检测
重组质粒pPIC9K/tps1全序列测定结果:
TGAAGCTTACGTAGAATTCATGTCCACCCCGGCAGAGACTCAGCAAGAGGCTGCTGCGCCCGGCCGTCTT CTGCTACTCTCGAACCGTCTGCTCATTGGCAGCTATACCTTTTCCATGTCCTCTGGTGGCCTTGTGACTGGCCTGA GCGGTCTGAGCAAGACCACCAGCTTCCAGTGGTGCGGATGGCCCGGCCTTGAGGTACCCGAGAACGAGGTCGA TGGCATGAAGAAACGCCTCAAAGCAGAGTACGGCGCGCATCCCGTCTTTATCGACGATGAGCTCGCTGACCGCC ACTACAATGGGTTTGCCAATTCTATCCTCTGGCCGTTGTTCCACTATCACCCCGGTGAAATCACCTTTGACGAGT CTGCCTGGGTCGCCTACCAAGAAGTCAACCGCCTCTTTGCGCAGACTGTCATCAAAGATGTTCAAGACGGTGAT CTAATCTGGGTGCACGACTACCATCTGATGCTTCTGCCCCAGATGTTGCGCGAGGAAATTGGCAACTCCAAGAA GAACGTCAAGATCGGCTTCTTTTTGCACACGCCATTTCCAAGTAGCGAAATTTACCGCATTCTGCCTGTCCGCGA GCCTCTTCTCACAGGTCTTCTCGATTGCGACCTGATTGGCTTCCACACATACGACTACGCGCGCCACTTCCTTAGT AGTTGCTCGCGCATACTCGGCACCCCCACCACCCCCAACGGTGTCGATTGGAATGGCCGCTTCGTGACTGTCGG GGCGTTTCCTATTGGAATAGACCCCGAAAAGTTTGTCGAGGGGTTGAAGCAACCCAAGGTGCAGGCGCGCATC GCAGCTCTGAAACGCAAGTTTGAAGGCGTGAAGCTCATTGTCGGCGTCGATCGTCTCGACTACATCAAGGGTGT GCCGCAAAAACTTCATGCCCTCGAGGTTTTCCTCACGGAGCACCCCGAGTGGATTGGCAAGATTGTCCTCGTCC AGGTTGCCGTGCCCTCACGCCAGGATGTCGAAGAGTACCAGAACCTGCGTGCCGTTGTCAACGAGCTCGTCGGC CGCATCAACGGCAAGTTTGGCACCATTGAATTTATGCCCATCCACTTTTTGCACCAGTCCGTGTCCTTTGACGAG CTGACGGCGCTCTACGCTGTGTCGGACGTGTGCCTGGTGTCTTCCACTCGTGATGGCATGAACCTCGTCTCGTAC GAGTACATTGCCACGCAGCGCGAGAAGCACGGCGTCATGATCCTCAGCGAGTTTACCGGCGCCGCGCAGTCGC TCAACGGCAGTCTGATTGTTAACCCCTGGAACACGGAGGAGCTAGCCAACGCCATCCACGACGCCGTGACCAT GAGCCCCGAGCAGCGCGCCGCCAACTACAGCAAGCTCGAGCGCTATGTCTTCAAGTACACGAGCGCTTGGTGG GGGGCCACCTTTGTCGCCGAGATGACGCGCCTCAGCAACGAGGAGTCGCAGGCAAAGACGATTCGCAATGTCT CGGGTGCCGTGGTCAGCTCGGGCCAAAAGGCACTGACAGAAGAGCCCGAGAACAAGGAGCCGGAGGCGAATC AAGCGGAATGAGCGGCCGCGAATTAATTCGCCTTAGACATGACT
两个方框是使用的限制性酶切位点,阴影部分之间是插入的TPS1基因序列。限制性酶切位点之外,是原始质粒pPIC9K的序列。
3 讨论
由于巴斯德毕赤酵母没有稳定的附加体质粒,因此需要使用整合型质粒作为外源插入基因的表达载体。其表达载体通常包括AOX1启动子(5’AOX1)、多克隆位点、AOX1转录终止子(AOXt)、组氨酸脱氢酶基因his4、3’AOX1以及来自大肠杆菌质粒pBR322的大肠杆菌复制序列。而整合位点一般位于HIS4区或AOX1区。巴斯德毕赤酵母用来表达外源基因,具有杂蛋白少、遗传稳定等优点[5]。它能对插入基因编码蛋白进行翻译后修饰,并分泌至细胞外[6-7]。其表达蛋白能够被糖基化,类似于人类蛋白质[8],是一种较为理想的真核蛋白表达系统。
本研究针对球孢白僵菌的tps1基因,构建了其真核表达载体pPIC9K/tps1。通过PCR鉴定和测序表明,该重组质粒包含了球孢白僵菌tps1基因的全长cDNA序列。并且Bbtps1基因被正确地插入到了pPIC9K质粒的表达框中。因此,该重组质粒可以直接用于tps1基因的酵母表达。
[1]洪洄,王冬梅.应用酵母进行基因表达的研究进展[J].生物学通报,1999,34(7):12-14.
[2]CereghinoJL,CreggJM.Heterologous protein expression in the methylotrophic yeast Pichia pastoris[J].FEMSMicrobiology Reviews,2000,24:45-66.
[3]Thor D,XiongS,OrazemCC,et al.Cloningand characterization ofPichia pastoris MET2 gene as a selectable marker[J].FEMS Yeast Research,2005,5(10):935-942.
[4]Sambrook J,Russell DW.Molecular Cloning[M].NewYork:Cold SpringHarbor LaboratoryPress,2001.
[5]Romanos M.Advances in the use ofPichia pastoris for high level gene expression[J].Current Opinion in Biotechnology,1995,6(5):527-533.
[6]HongF,Meinander NQ,Jonsson LJ.Fermentation strategies for improved heterologous expression oflaccase in Pichia pastoris[J].Biotechnologyand Bioengineering,2002,79(4):438-449.
[7]Sreekrishna K,Brankamp RG,Kropp KE,et al.Strategies for optimal synthesis and secretion ofheterologous proteins in the methylotrophic yeast Pichia pastoris[J].Gene,1997,190(1):55-62.
[8]Duman JG,Miele RG,HongL,et al.O-Mannosylation ofPichia pastoris cellular and recombinant proteins[J].Biotechnologyand Applied Biochemistry,1998,28(1):39-45.
