肉桂酰胺类化合物抗癌活性研究进展
2013-04-20徐长亮王菊仙白晓光王玉成
徐长亮,王菊仙,白晓光,王玉成
针对肉桂酰胺类化合物的药物研发已有几十年的历史,国内外文献对其药理活性有许多报道,其中大部分都集中在抗惊厥[1]、扩张血管[2]、杀虫[3]、抗诱变[4]、治疗糖尿病[5]、抗抑郁[6]以及消炎[7]等方面。近二十年,肉桂酰胺类化合物抗肿瘤活性的报道逐年增多,本文就此展开综述,希望能为肉桂酰胺类药物的研发提供参考。
肉桂酰胺及其相关化合物在自然界中广泛存在,形成了一系列具有抗癌活性的物质[8],具体分为以下几类:
1 α-氰基肉桂酰胺类
Shiraishi 等[9]在筛选酪氨酸激酶抑制剂时发现,3-氰 基-4-羟基肉桂酰胺和 3,5-二叔丁基-α-氰基-4-羟基肉桂酰胺具有潜在的活性,于是在此结构基础上,设计合成了一系列 α-氰基肉桂酰胺类化合物(1a ~ 1w),结构如表 1 所示。对酪氨酸激酶的抑制活性测试结果显示,将 3,5-二叔丁 基-α-氰基-4-羟基肉桂酰胺的 3、5 位用苯基、乙氧基和苯硫基甲基取代后得到的化合物 1b 和 1f 活性最佳,对表皮鳞状癌 A-431 细胞的 IC50均达到 0.37 μmol/L。构效关系研究显示,苯环上 R2位的羟基及其与酰胺键之间的碳碳双键对活性至关重要,并且苯环上 R1和 R3位是疏水性基团取代时,可增强活性。由于酪氨酸激酶抑制剂可以抑制酪氨酸激酶在细胞的增殖、变异、致癌以及肿瘤进程的控制中所起到的关键性作用,进而发挥肿瘤预防和化疗的功效,所以化合物 1b 和 1f 具有潜在的抗癌活性,可以作为抗肿瘤药物来开发。但由于某些未知原因,未见此类化合物抗肿瘤活性的后续报道。
2 2-甲基肉桂酰胺
2-甲基肉桂酰胺是 Welch 等[10]从灰藤黄链霉菌(Streptomyces griseoluteus)发酵的啤酒液中分离得到的,结构如图 1 所示。该化合物在抑制癌细胞侵袭和转移方面效果显著。当使用非毒性剂量对恶性人黑色素瘤细胞(C8161 和 A375M)进行预处理时,发现瘤细胞的侵袭出现了对剂量和时间依赖的可逆性下降(IC50= 12.5 μg/ml);而当在静脉注射前将肿瘤细胞进行与前述同样的预处理时,发现其对肺的浸染被显著抑制(P < 0.05)。2-甲基肉桂酰胺对癌细胞的作用机制尚未明确,需要后续的研究加以揭示。
3 氮芥肉桂酰胺类
研究发现,偏端霉素的肉桂氮芥类衍生物(3a)和他莫司汀(3b)对白血病具有良好的治疗效果,并且 3a 的活性明显高于 3b。由于两者分子中的脒基部分具有强碱性,在任何生物环境下都能够发生质子化,所以在 DNA 结合以及细胞或组织的生物利用度方面都可能发挥重要的作用。另外在 R1基团处,3a 较 3b 多一个碳碳双键,延长的共轭键可能对活性提升具有一定贡献。将 3a 和 3b 分子中的 3 个吡咯环换成吡唑环得到化合物 3c 和 3d,其中 3d 的活性要比 3c 强 20 倍,与 3a 基本相当[11-12]。将 3d 分子中脒基部分用不同性质的碱性或非碱性脒基片段取代后得到化合物 3e ~ 3i,结构如图 2 和表 2 所示。其中除了 3e 的活性与 3d 保持相当之外,其余化合物的活性均出现了一定的下降,说明脒基的存在对活性具有重要贡献。 Baraldi 等[13]的研究表明,化合物 3d ~ 3i 具有与富含 AT 序列的 DNA 选择性相互作用的能力。与其他报道的类似化合物一样,偏端霉素作为这些新分子与 DNA 特异性绑定的载体,将氮芥片段定位于一个非常适于下一步烷基化的位置,因而此类化合物具有高效以及特异性的抗癌作用。
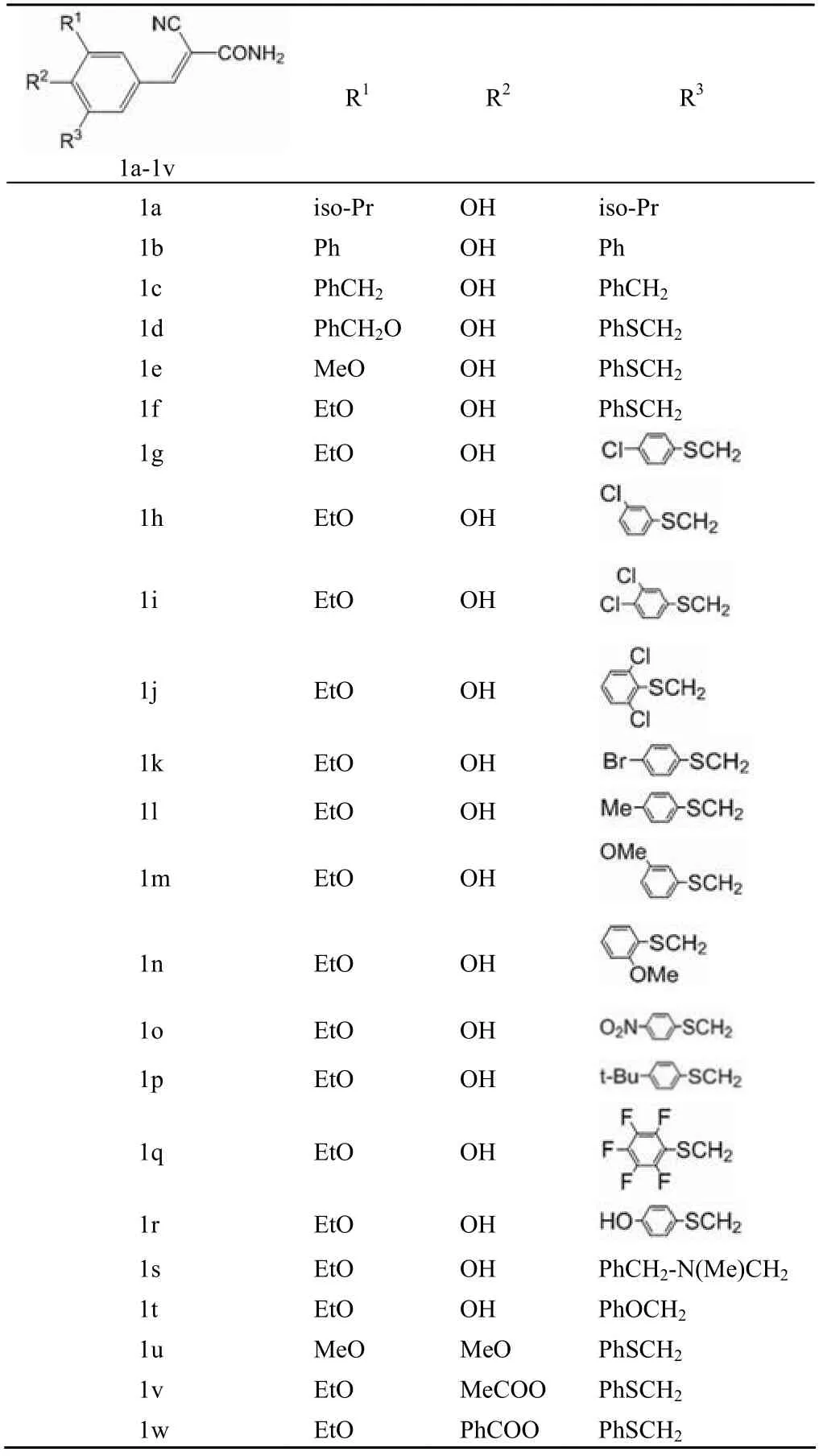
表1 化合物 1a ~ 1w 的结构
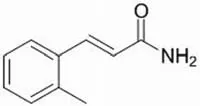
图1 2-甲基肉桂酰胺的结构
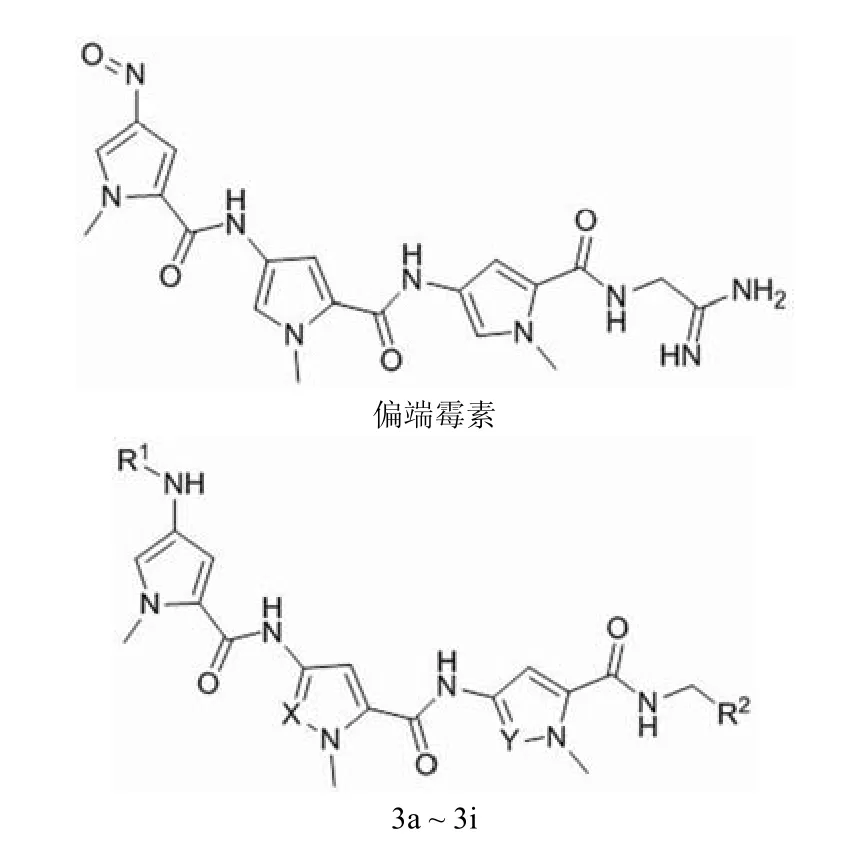
图2 偏端霉素和化合物 3a ~ 3i 的结构
4 肉桂酰四氢吡咯类
Nagamura 等[14]报道,将 duocarmycin A 的吡咯环部分与对硝基苯酚肉桂酸酯经 NaH 处理得化合物 4a ~ 4k,吡咯环中的环丙基用氢溴酸打开,再引入氮取代氨基甲酸酯基团可得到化合物 4l ~ 4s,结构如图 3、表 3 和表 4 所示。对这些肉桂酰四氢吡咯类化合物的抗癌活性评价发现,化合物 4a 和 4b 具有很强的细胞生长抑制活性;化合物 4c ~ 4k 的邻位取代对活性没有多少影响,但是对水溶性的提高有很大帮助。这些结果促成了化合物 4l ~ 4s 的合成。其中,化合物 4l ~ 4p 具有抑制小鼠 S180 实体瘤细胞的活性。与 4l ~ 4p 相比,4q ~ 4s 在甲氧基的邻位引入了亲水性基团,水溶性有所提高,但活性比前者低。这表明类似化合物中富电子基团的存在可能会导致细胞通透性的降低,进而影响抗癌作用。
基质金属蛋白酶类(MMPs)是一类涉及众多生理以及病理过程的水解酶,其过度激活将导致包括肿瘤在内的许多疾病的发生。在 MMPs 中,明胶酶类是一类重要的酶,其在恶性组织中被发现大量上调,且在癌变及血管生成中也有表达。另外,明胶酶类几乎涉及肿瘤进程的每一个阶段[15]。因此,寻找此类酶的抑制剂成为研究热点。Zhang 等[16]在 L-羟脯氨酸结构的基础上合成了一系列新的肉桂酰四氢吡咯类化合物 4t ~ 4v,结构如图 4 和表 5 所示。经白明胶酶(MMP-2 & 9)和氨肽酶N(APN)抑制活性测试,在吡咯环 C4 位连有芳环侧链的化合物 4t ~ 4v 均为良好的白明胶酶抑制剂。但与对 MMP-2 的活性相比,大部分化合物对 APN 的活性较差。
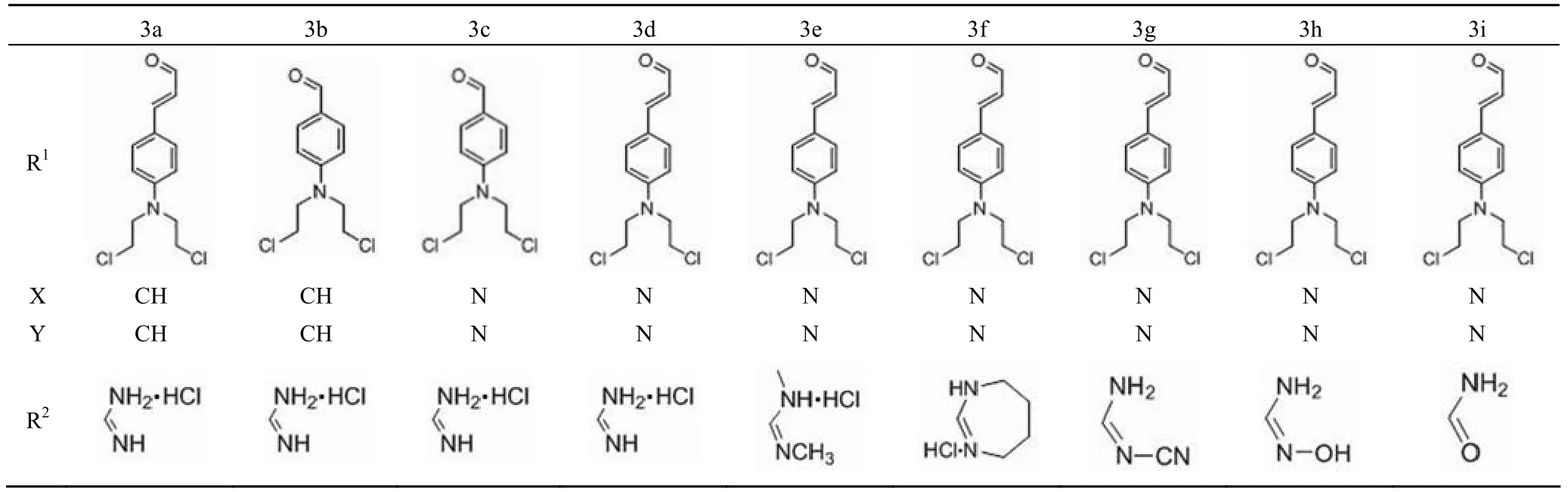
表2 化合物 3a ~ 3i
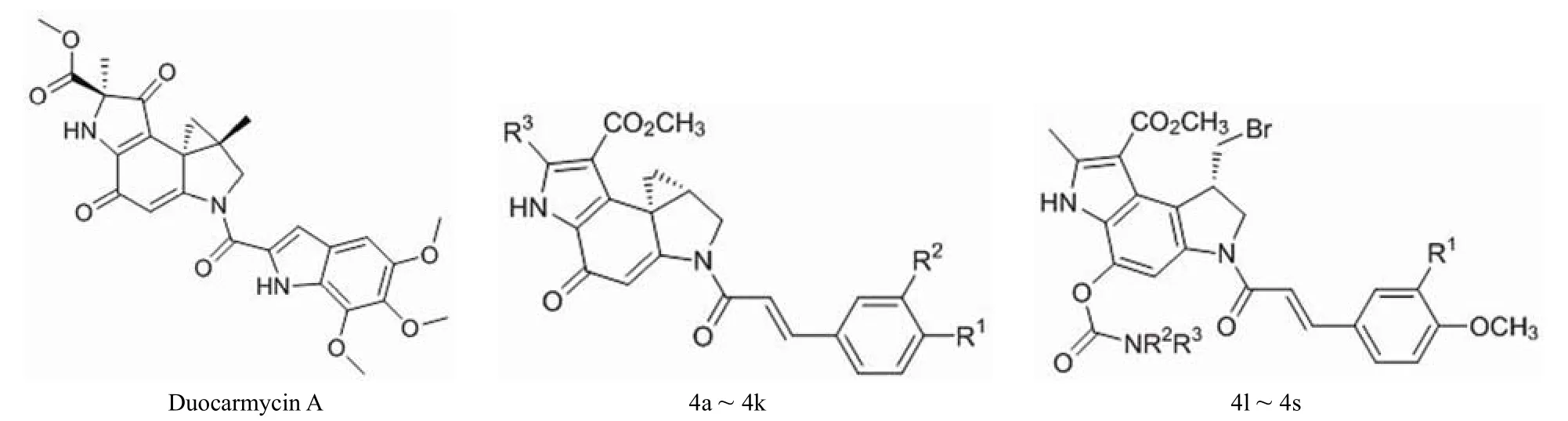
图3 Duocarmycin A 和化合物 4a ~ 4s 的结构
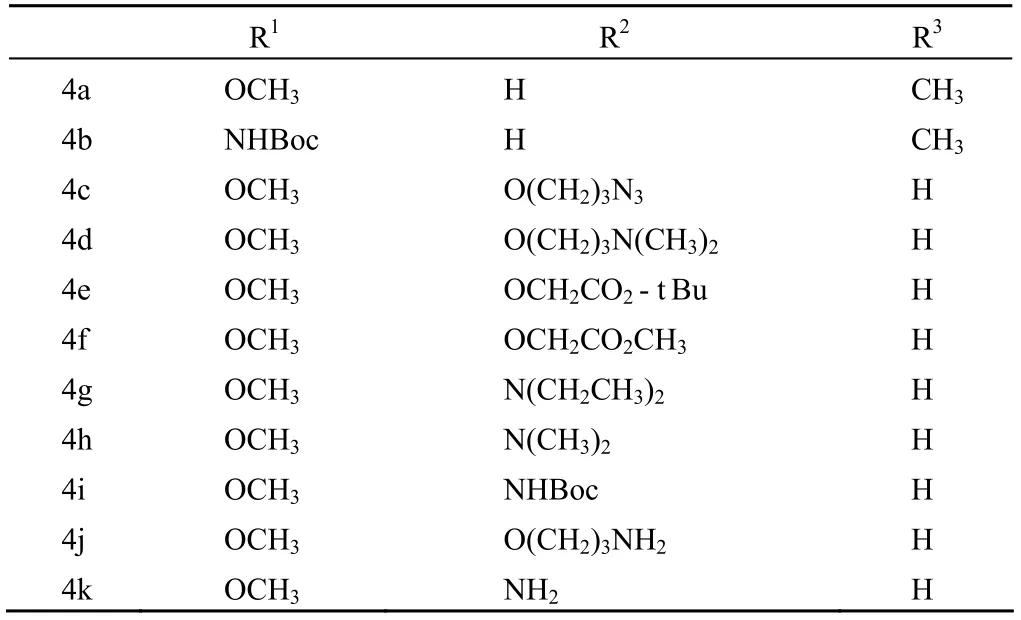
表3 化合物 4a ~ 4k
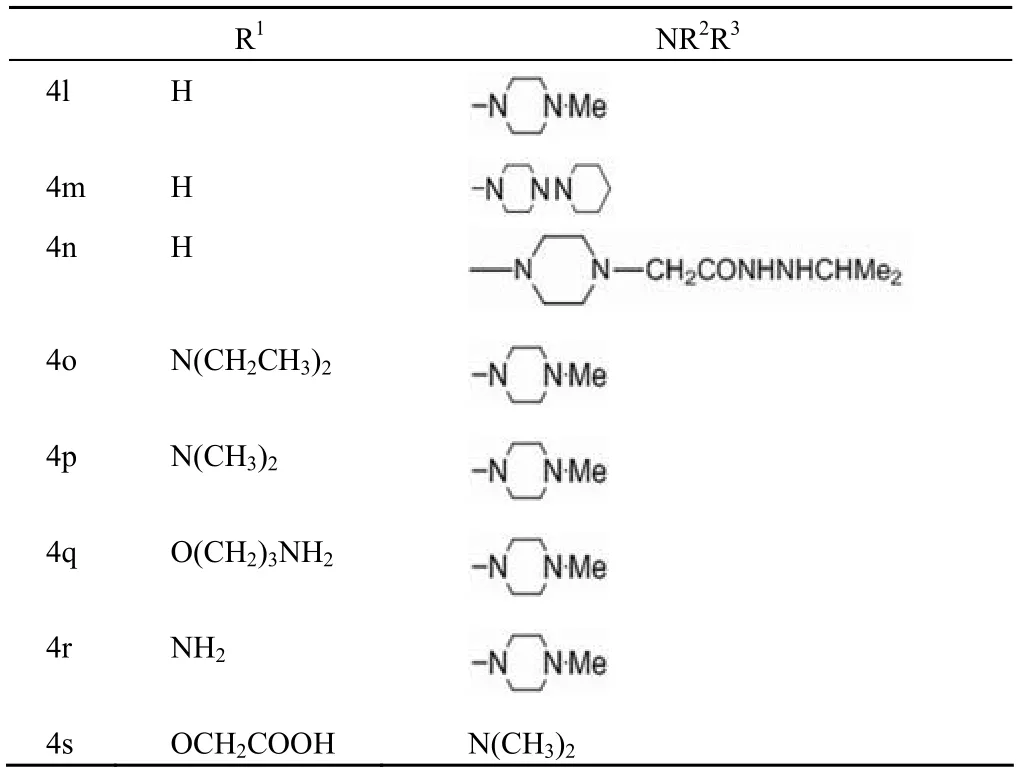
表4 化合物 4l ~ 4s
5 肉桂酰胺
Jiang 和 Zhen[17]对肉桂酰胺(CNM)(图 5)的抗癌活性进行了研究,发现其对人口腔表皮样癌 KB 细胞、人肝癌 BEL-7420 细胞以及人纤维肉瘤 HT-1080 细胞的 IC50值为 1.29 ~ 1.94 mmol/L;对人胎儿肺 2BS 细胞的 IC50值为 4.33 mmol/L;当以 50 ~ 150 mg/kg 的剂量对小鼠给药(静注或口服)时,显示了中等的抑制肿瘤活性;静注或口服肉桂酰胺(150 mg/kg)对移植的小鼠肝癌细胞 H22 的抑制率分别达到 48.8% 和 40.5%;100 mg/kg 的肉桂酰胺对小鼠结肠癌细胞 CT26 以及 Lewis 肺癌细胞的抑制率分别为 39.0% 和 53.9%;在 Lewis 肺癌模型中,100 mg/kg的肉桂酰胺静注也使癌细胞的转移降低了 59.1%;明胶酶谱法显示,肉桂酰胺能以浓度依赖的方式降低 HT-1080 肿瘤 细胞条件培养液中 MMP-2 的水平。上述结果说明,肉桂酰胺是一种作用于 MMPs 的低毒抗肿瘤化合物,可作为抗肿瘤药物开发的先导物。
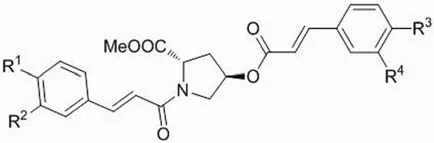
图4 化合物 4t ~ 4v 的结构
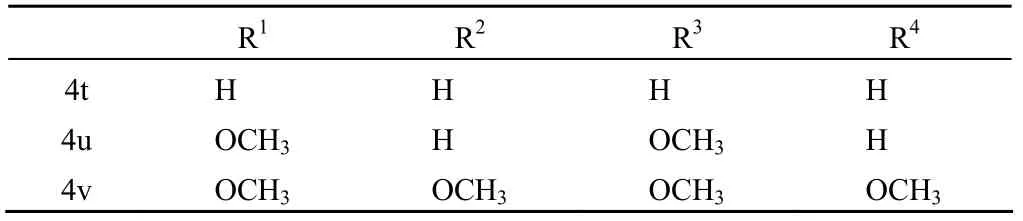
表5 化合物 4t ~ 4v
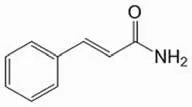
图5 肉桂酰胺的结构
6 N-甲基肉桂酰胺丁酸甲酯
组蛋白的乙酰化能够促进 DNA 的转录,进而强化基因的表达。但是组蛋白去乙酰化酶(HDACs)却能够逆转这个过程,抑制基因的表达。HDACs 在细胞中调节细胞周期抑制因子 p21 的表达和肿瘤抑制因子 p53 的活性,在肿瘤的发生中起着重要的作用,以其为靶点的小分子抑制剂的设计与合成也成为近年来抗肿瘤药物研发的热点之一。Lan-Hargest 和 Weich[18]的研究显示,N-甲基肉桂酰胺丁酸甲酯(图 6)可与组蛋白去乙酰化酶活性中心的锌离子结合并形成螯合物,能有效地抑制组蛋白去乙酰化酶的活性,是一种优良的组蛋白去乙酰化酶抑制剂。然而通过实验发现,N-甲基肉桂酰胺丁酸甲酯对人乳腺癌 MCF-7 细胞和前列腺癌 PC-3 细胞的抑制活性并不高,IC50只有 20 μmol/L。
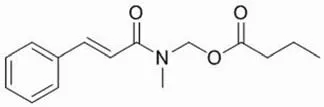
图6 N-甲基肉桂酰胺丁酸甲酯的结构
7 肉桂酰羟胺类
为寻找组蛋白去乙酰化酶的小分子抑制剂,焦杰等[19]设计合成了 11 种肉桂酰胺类化合物 7a ~ 7k,结构如图 7 所示,并对其 HDACs 抑制活性进行了测试。结果表明,11 种化合物的酶抑制活性均不及阳性对照药 SAHA(商品名 Zolinza,IC50= 1.28 μmol/L),其中以化合物 7a、7b、 7e 和 7k 活性较好,IC50= 2 ~ 15 μmol/L。
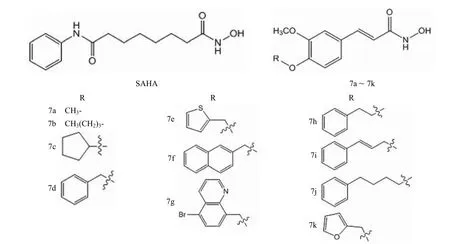
图7 SAHA 与化合物 7a ~ 7k 的结构
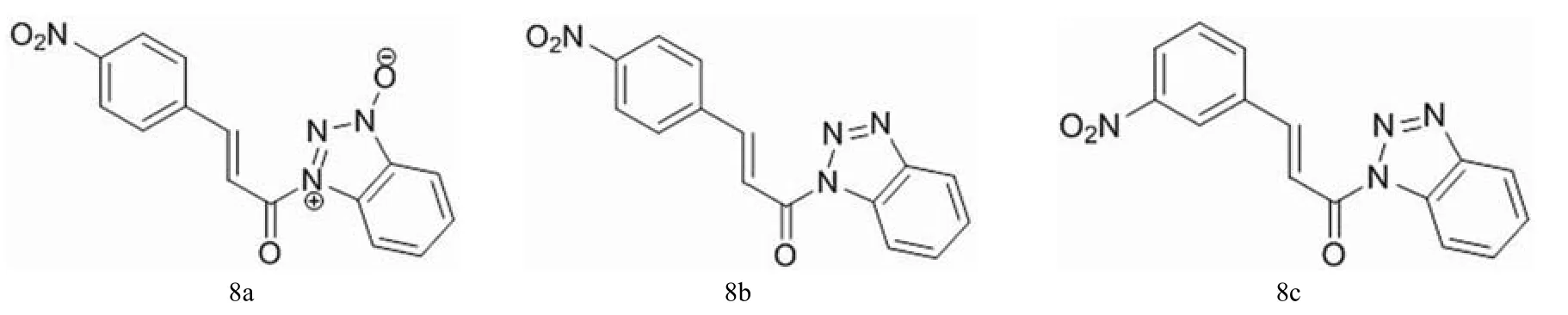
图8 化合物 8a ~ 8c 的结构
8 肉桂酰三氮唑类
转谷酰胺酶(TGases)是一类钙依赖酶,可以通过形成侧链间的 γ-谷酰基-ε-赖氨酸来催化特定蛋白的分子间偶联。高活性的 TGases 会导致包括银屑病、免疫系统疾病以及肿瘤转移等在内的许多生理疾病的发生。为寻找其抑制剂,Pardin 等[20]合成了肉桂酰三氮唑类化合物 8a ~ 8c,结构如图 8 所示。这些化合物都是 TG2 的可逆抑制剂,其中以 8b 和 8c 的活性最佳,IC50值分别为 43 mmol/L 和 24 mmol/L。大部分有效抑制剂分子的对位取代基中都包含 sp2 杂化的氧原子,这样既保持了肉桂双键的距离,又维持了共轭;而其中酰化的杂环则可作为氢键的受体。这些抑制剂可能是绑定在酰基供体结合位点的疏水凹槽内发挥作用的。苯并三唑是很好的离去基团,所以这些分子可以作为良好的酰化试剂。
9 肉桂酰嘧啶类
酚氨嘧啶(2-phenylaminopyrimidine,PAP)衍生物 STI-571 是一种有效并具有相对选择性的 Bcr-Abl 酪氨酸激酶抑制剂,对处于慢性期的慢性髓性白血病患者具有很好的疗效,但 Bcr-Abl 激酶域的突变已被证实为对 STI-571 耐药的主要原因之一。受此启发,Chang 等[21]开发了一系列新的 PAP 衍生物 9a ~ 9f,结构见图 9 和表 6。抗癌活性测试结果显示,与 STI-571 相比,9b 和 9c 的活性明显提高,而 9d 则基本持平,其他化合物的活性出现不同程度 的降低。可见将 STI-571 的苯酰部分换成苯环上被合适的吸电子取代基取代的肉桂酰后,活性得到了提升,说明这种不饱和芳香酮结构对此类抑制剂分子的活性提高有益。
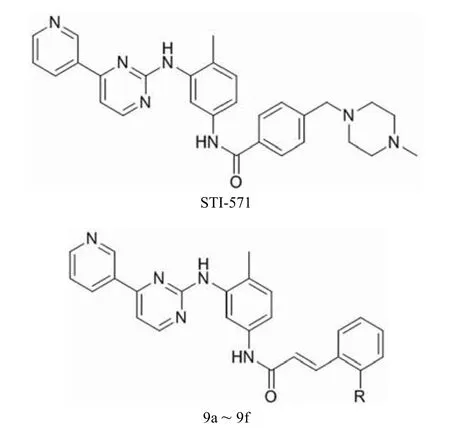
图9 STI-571 和化合物 9a ~ 9f 的结构
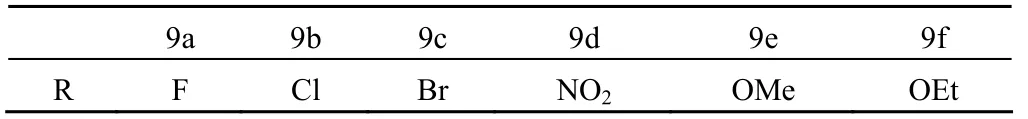
表6 化合物 9a ~ 9f
10 肉桂酰哌嗪类
Srivastava 等[22]将阿魏酸与哌嗪类衍生物以及 [4-(2-溴烷氧基)-5-甲氧基-2-硝基苯基-(S)-2-(二乙硫甲基)四氢吡咯]-1-甲酮对接,合成了一些肉桂酰哌嗪类化合物 10a1 ~ z9,结构见图 10。由阿魏酸出发,将其与不同的哌嗪对接,得到化合物 10a1 ~ 10a3 和 10w。在此基础上,将其苯环对位的羟基与 [4-(2-溴烷氧基)-5-甲氧基-2-硝基苯基-(S)- 2-(二乙硫甲基)四氢吡咯]-1-甲酮对接,得到化合物 10b1 ~ 10j9 以及 10x1 ~ 10x9;而将其分子中硝基还原为氨基则得到化合物 10k1 ~ 10s9 和 10y1 ~ 10y9,然后合环又可得化合物 10t1 ~ 10v9 和 10z1 ~ 10z9。对这些化合物抗癌活性 进行了评价,包括 60 种不同的人类肿瘤细胞,如 Hop-62、SiHa、MCF-7、Zr-75-1 以及 Colo205 等。结果显示,化合物 10v2 活性最好(GI50= 0.02 ~ 3.39 μmol/L),并且所有的化合物对癌细胞均有毒性,同时对 DNA 的绑定能力也很显著。
11 肉桂异羟肟酸类
为提升氨肽酶抑制剂 bestatin 对成人急性白血病的临床治疗效果,Liu 等[23]设计合成了一系列 N-肉桂酰-L-天冬氨酸及其异羟肟酸衍生物 11a ~ 11f,结构如图 11 所示。由 L-天冬氨酸出发,将其酯化后与肉桂酰连接得化合物 11a;将 11a 水解得对应的酸 11b;11b 在醋酸酐中加热脱水得对应的酸酐 11c;再用正丁胺处理则得化合物 11d;然后在盐酸中用羟胺处理 11d 得到化合物 11e。这些化合物对氨肽酶的抑制活性测试显示,化合物 11e 的活性最好 [IC50=(11.1 ± 0.9)μmol/L];另外,与 bestatin 相比,11e 对 HL-60 细胞(多药耐药白血病)的抗增殖活性更加优异。

图10 阿魏酸和化合物 10a1 ~ 10z9 的结构(n = 1 ~ 9)
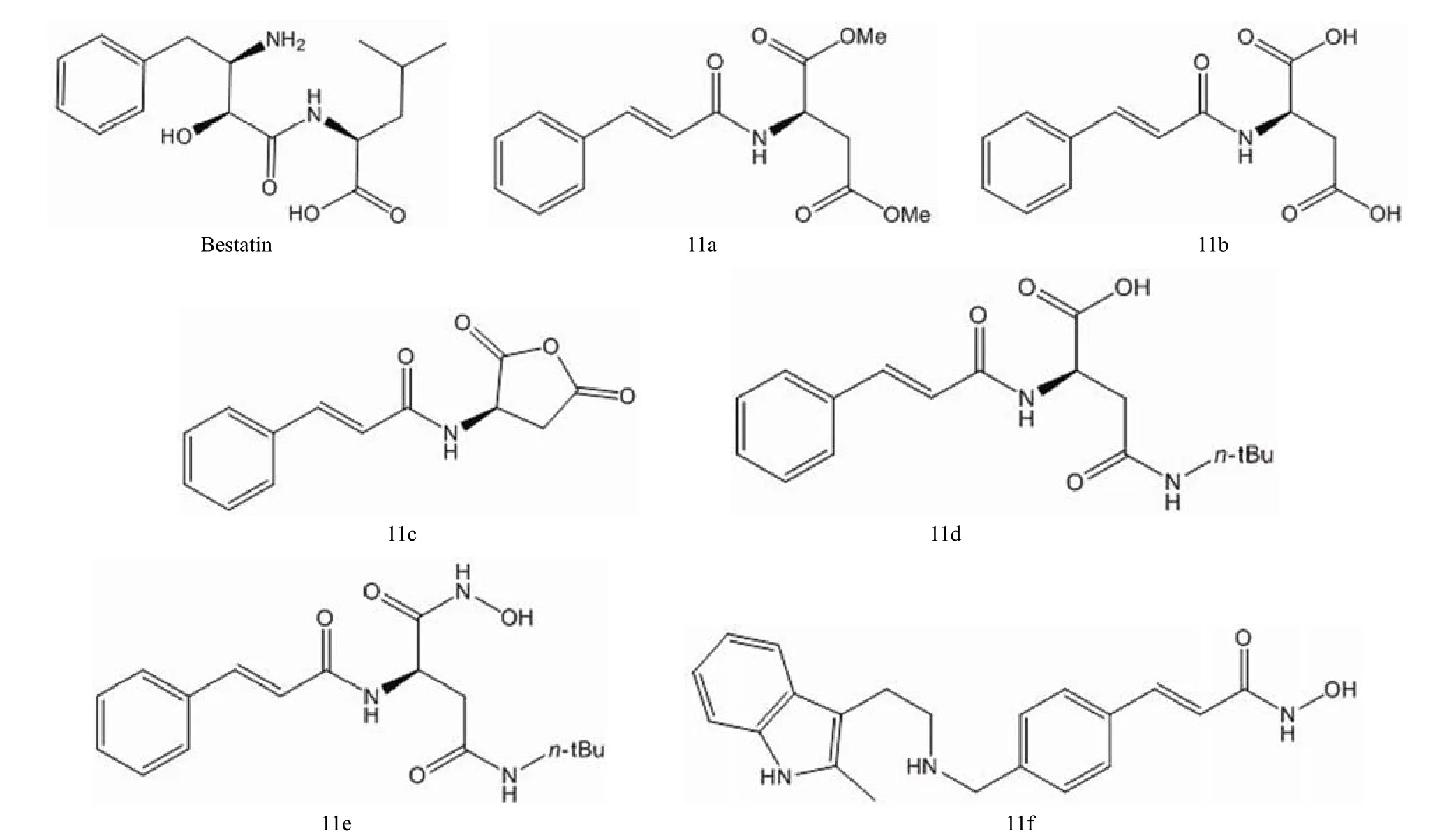
图11 Bestatin 和化合物 11a ~ 11f 的结构
化合物 11f 的结构与上述几个化合物差异较大,是一种新的肉桂异羟肟酸类组蛋白脱乙酰化酶抑制剂,可诱导人类慢性髓细胞性白血病急变期细胞 K562 和急性白血病细胞 MV4-11 的凋亡。
12 肉桂酰苯胺类衍生物
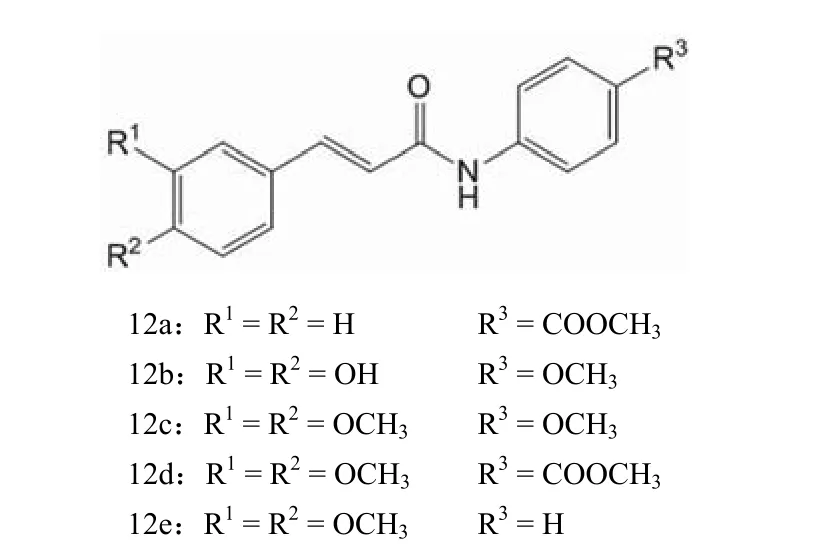
图12 化合物 12a ~ 12e 的结构
姜集苗等[24]合成了 5 种类似查尔酮结构的肉桂酰苯胺类化合物 12a ~ 12e,并采用 MTT 比色实验对其抗癌活性进行了筛选。实验以肉桂酸为阳性对照物,结果显示,化合物 12a ~ 12e 均有一定的抑癌活性,其中对人子宫颈癌细胞系 HeLa 细胞和乳腺细胞系 MDA-MB-435 细胞的抑制活性以 12e 最高,IC50分别为 42.38 μg/ml 和 10.05 μg/ml,而对人肺癌细胞系 H460 细胞的抑制活性则以 12b 最佳,IC50为34.98 μg/ml。 从上述研究结果来看,大部分合成或分离得到的肉桂酰胺类化合物的抗癌活性欠佳。原因在于大部分的研究系统性不强,或是从菌株发酵液中分离纯化后进行抗癌活性筛选,或是仿照经典分子或已有药物分子,设计合成后进行活性筛选,并没有对此类化合物进行系统的构效关系研究。由于肉桂酰部分具有 3 个优秀的反应位点,可以进行苯环上的取代、α,β-不饱和双键的加成以及羧基部分的变化等,从而衍生出数量庞大的化合物。通过对比这些化合物中苯环上取代基性质的不同、数量的不同和取代位置的不同,碳碳双键是否加成,加成取代基位置和种类的不同,还有羰基部分的改变,可以揭示此类化合物结构中不同部位对生物活性的作用。笔者认为,在对构效关系研究较为全面的基础上,一定可以找到活性更好的肉桂酰胺类抑癌小分子化合物。
[1] Guan LP, Wei CX, Deng XQ, et al. Synthesis and anticonvulsant activity of N-(2-hydroxyethyl) cinnamamide derivatives. Eur J Med Chem, 2009, 44(9):3654-3657.
[2] Mou LY. 1. Synthesis and vasodilating activity of α-phenylcinnamides, 2. Synthesis and quantitative structivity activity relationship of PAF-receptor antagonists, aromatic aminoketones, 3. Total synthesis of Chrysanthemum indicum L. Beijing: Chinese Academy of Medical Sciences & Peking Union Medical College, 1998. (in Chinese) 牟丽媛. 一、α-苯基取代肉桂酰胺的合成及其扩血管活性研究 二、PAF 受体拮抗剂—芳氨酮类化合物的合成及构效关系研究 三、天然产物野菊花醇的全合成. 北京: 中国医学科学院&北京协和医学院, 1998.
[3] Kong JO, Lee SM, Moon YS, et al. Nematicidal activity of cassia and cinnamon oil compounds and related compounds toward bursaphelenchus xylophilus (nematoda parasitaphelenchidae). J Nematol, 2007, 39(1): 31-36.
[4] Motohashi N, Ashihara Y, Yamagami C, et al. Structure-antimutagenic activity relationships of benzalacetone derivatives against UV-induced mutagenesis in E. coli WP2uvrA and gamma-induced mutagenesis in Salmonella typhimurium TA2638. Mutat Res, 2001, 474(1-2):113- 120.
[5] Nie W, Luo JG, Wang XB, et al. Synthesis of new α-glucosidase inhibitors based on oleanolic acid incorporating cinnamic amides. Chem Pharm Bull (Tokyo), 2011, 59(8):1051-1056.
[6] Deng XQ, Wu D, Wei CX, et al. Synthesis and antidepressant-like action of N-(2-hydroxyethyl) cinnamamide derivatives in mice. Med Chem Res, 2011, 20(8):1273-1279.
[7] Ronad PM, Hunashal RD, Darbhamalla S, et al. Synthesis and evaluation of anti-inflammatory and analgesic activities of a novel series of substituted-N-(4-methyl-2-oxo-2H-chromen-7-yl) benzamides. Arzneimittelforschung, 2008, 58(12):641-646.
[8] De P, Baltas M, Bedos-Belval F. Cinnamic acid derivatives as anticancer agents-a review. Curr Med Chem, 2011, 18(11):1672-1703.
[9] Shiraishi T, Kameyama K, Imai N, et al. Specific inhibitors of tyrosine-specific protein Kinase. I. Synthesis and inhibitory activities of alpha-cyanocinnamamides. Chem Pharm Bull (Tokyo), 1988, 36(3):974-981.
[10] Welch DR, Harper DE, Yohem KH. U-77, 863: a novel cinnamide isolated from Streptomyces griseoluteus that inhibits cancer invasion and metastasis. Clin Exp Mestastasis, 1993, 11(2):201-212.
[11] Cozzi P, Beria I, Biasoli G, et al. Novel phenyl nitrogen mustard and half-mustard derivatives of amidino-modified distamycin. Bioorg Med Chem Lett, 1997, 7(23):2979-2984.
[12] Cozzi P, Beria I, Biasoli G, et al. Novel phenyl nitrogen mustard and half-mustard derivatives of distamycin A. Bioorg Med Chem Lett, 1997, 7(23):2985-2990.
[13] Baraldi PG, Beria I, Cozzi P, et al. Cinnamoyl nitrogen mustard derivatives of pyrazole analogues of tallimustine modified at the amidino moiety: design, synthesis, molecular modeling and antitumor activity studies. Bioorg Med Chem, 2004, 12(14):3911-3921.
[14] Nagamura S, Asai A, Amishiro N, et al. Synthesis and antitumor activity of duocarmycin derivatives: A-ring pyrrole compounds bearing cinnamoyl groups. J Med Chem, 1997, 40(6):972-979.
[15] Vargová V, Pytliak M, Mechírová V. Matrix metalloproteinases. EXS, 2012, 103:1-33.
[16] Zhang L, Zhang J, Fang H, et al. Design, synthesis and preliminary evaluation of new cinnamoyl pyrrolidine derivatives as potent gelatinase inhibitors. Bioorg Med Chem, 2006, 14(24):8286-8294.
[17] Jiang X, Zhen Y. Cinnamamide, an antitumor agent with low cytotoxicity acting on matrix metalloproteinase. Anticancer Drugs, 2000, 11(1):49-54.
[18] Lan-Hargest HY, Weich NL. Preparation of dicarbonyl compounds as inhibitors of histone deacetylase: Eur, EP 1216986. 2000-12-21.
[19] Jiao J, Fang H, Zhu HW, et al. Synthesis and activity study of a new series of histone deacetylases (HDACs) inhibitors of N- hydroxycinnamamide derivatives. Chin J Med Chem, 2008, 18(4): 250-253. (in Chinese) 焦杰, 方浩, 朱华伟, 等. 一组新的N-羟基肉桂酰胺类组蛋白去乙酰化酶抑制剂的合成和初步活性研究. 中国药物化学杂志, 2008, 18(4):250-253.
[20] Pardin C, Pelletier JN, Lubelle WD, et al. Cinnamoyl inhibitors of tissue transglutaminase. J Org Chem, 2008, 73(15):5766-5775.
[21] Chang S, Yin SL, Wang J, et al. Design and synthesis of novel 2-phenylaminopyrimidine (PAP) derivatives and their antiproliferative effects in human chronic myeloid leukemia cells. Molecules, 2009, 14(10):4166-4179.
[22] Srivastava V, Srivastava AM, Tiwari AK, et al. Disubstituted 4(3H) quinazolones: a novel class of antitumor agents. Chem Biol Drug Des, 2009, 74(3):297-301.
[23] Liu Y, Shang L, Fang H, et al. Design, synthesis, and preliminary studies of the activity of novel derivatives of N-cinnamoyl-l-aspartic acid as inhibitors of aminopeptidase N/CD13. Bioorg Med Chem, 2009, 17(20):7398-7404.
[24] Jiang JM, Hu GH, Zuo MM, et al. Study on the synthesis and anti-tumor activity of novel chalcone analogues. Chem Res Appl, 2010, 22(11):1045-1048. (in Chinese) 姜集苗, 胡国辉, 左明明, 等. 查儿酮类似物的合成及抗肿瘤活性研究. 化学研究与应用, 2010, 22(11):1045-1048.
