烟煤基活性炭的制备及脱除甲基橙性能
2013-03-03吴永红沈国良赵丹丹党晓龙
吴永红,张 兵,沈国良,赵丹丹,党晓龙
(沈阳工业大学石油化工学院,辽宁辽阳111003)
活性炭 (AC)因具有高度发达孔隙结构及特殊表面特性,已广泛用于环保、化工、石油、食品、冶金、药物、军事等诸多重要领域[1-2]。目前,我国所产AC大都性能较差,缺乏国际市场竞争力。亟待开发高性能AC的制备新技术。以传统木材、果壳及农副产品为原料制备AC不仅受来源及品位限制,且得率低、吸附性能小[3-7]。煤因具有碳含量高、价格低优势,适于用作原料[8]。Kopac[9]以煤为前体,经脱灰、炭化及活化得到AC的比表面积为830.5 m2/g。Usmani等[10]将煤经ZnCl2活化得到AC的碘值与亚甲基蓝值分别为990 mg/g和205 mg/g,比表面积为942 m2/g。Hsu等[11]从KOH活化制得煤基AC的孔隙率达3300 m2/g。Ganan等[12]采用KOH-空气联合活化法从烟煤制备了高孔隙率AC。在国内,解强等开发了KOH、K2CO3与ZnCl2等活化剂及微波加热技术制备煤基AC,研究了其电化学性、吸附性、孔结构,并通过添加Fe3O4制备了便于回收的磁性AC[13-15]。邢宝林等[16-18]以NaOH和KOH为活化剂制备的煤基AC比表面积达24833215 m2/g。张传祥等[19]研究了KOH/NaOH混合活化剂的协同活化作用。
我国作为世界最大煤炭储量与生产国,为煤基AC的生产与发展奠定了基础。本文基于阜新烟煤开展了AC的制备,为发展以洁净消耗、防治污染的能源消耗模式,实现煤产业可持续发展战略奠定基础,有利于提高国际市场上的竞争力。
1 实验部分
1.1 原料
原料为阜新烟煤,按国家标准[20]测其工业分析指标为水分(Mad)4.498%,挥发分(Vad)27.96%,含碳量(Fcad)40.26%,灰分(Aad)27.29%。所用试剂均为市售,盐酸(36%38%),硫酸(95%98%),氢氟酸(40%),无水碳酸钠(99.8%),氯化锌(98%),氢氧化钾(82%),硫代硫酸钠 (99%),重铬酸钾(99.8%,),碘化钾(98%)、可溶性淀粉,及商品果壳基活性炭 (沈阳力诚试剂厂)。
1.2 实验过程
(1)煤的脱灰 向粒度为100目煤粉中加适量15%盐酸,在60℃保持1 h。过滤后,用蒸馏水洗至中性。再加适量25%氢氟酸,60℃保持2 h。过滤后洗至中性,经烘箱干燥去除水分。将经过上述脱灰与未脱灰的煤分别记为煤-D和煤-R。
(2)活性炭制备 将5 g脱灰煤粉与活化剂水溶液(KOH或ZnCl2)混合均匀,加热干燥至泥浆状。再放入炭化炉,在氮气氛围中以3℃/min速度升至600800℃,恒温一段时间(KOH与ZnCl2活化时的恒温时间分别为90 min和50 min)后,自然降至室温。将样品浸入3 mol/L盐酸,加热至沸腾15 min,再用蒸馏水洗涤至中性,过滤、干燥后得活性炭。从KOH与ZnCl2活化制得活性炭分别标记为AC-K与AC-Zn。
1.3 样品表征
采用HCT-1型热重分析仪以20℃/min升温速率,测定了空气氛围下样品从室温升到800℃的质量变化。经日本岛津XRD-7000型X射线衍射仪测定了样品的衍射谱图。通过美国Nicolet Nexus 470型ATR-FTIR光谱仪测定了样品表面官能团。经利曼有机元素分析仪EA3000测定了单位样品中有机元素C、H、O和N的量。分别采用国家标准规定的碘值[21]与四氯化碳 (乙醇)蒸汽吸附法[22]测定了样品吸附性及孔体积。
另外,通过脱除水中甲基橙的效果评价了样品吸附性能。将活性炭置于盛装甲基橙水溶液的磨口锥形瓶内,在水浴摇床上待吸附一定时间后,通过可见分光光度计测定吸附前后溶液吸光度。再由吸光度-浓度的标准曲线及物料衡算,获得活性炭的脱除率。
2 结果与讨论
2.1 脱灰对煤样品的影响
煤的灰分是由矿物质燃烧产生的,不仅影响活性炭机械强度,也会降低比表面积与孔体积,降低吸附性。因此,常需对原料煤脱灰,提高AC性能。本文烟煤经脱灰后,灰分(Aad)由27.29%显著降低至7.05%。
从XRD谱图(图1)可见,煤-R中4个衍射峰12.26°、20°、25.85°和36.26°,表明存在SiO2、Fe2O3、Al2O3、CaO、MgO等矿物质。煤-D谱图中,上述4峰基本消失,但仍可见20.88°、34.9°、39.42°和56.34°处衍射峰,说明矿物质仍有少量残存。
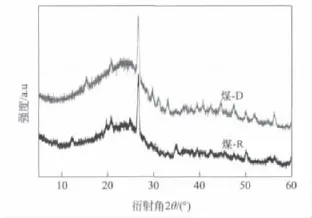
图1 脱灰前后煤样品的XRD谱图
红外谱图(图2)所示,煤-R在1033.72 cm-1处有强C—O键反射峰[17];而煤-D的此峰很微弱,说明脱灰后煤表面C—O官能团大多被脱除。另外,煤-R与煤-D中均含C—H键反射峰(1442 cm-1)。煤-R与煤-D相比,在29003600 cm-1间的—O—H键反射峰更强且峰形尖锐,说明脱灰后部分—O—H官能团也遭到破坏或脱除。
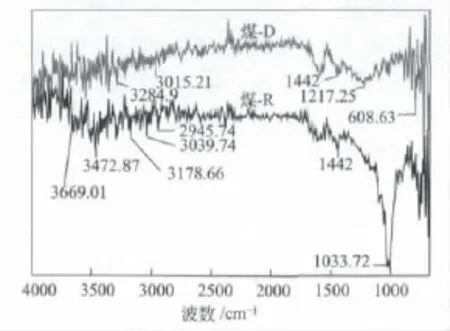
图2 脱灰前后煤的红外光谱图
从表1可看到,脱灰后N、C、H、和O等元素量都比脱灰前提高,说明灰分占到原煤样品中较大组成比例,脱灰显著提高了有机物组成的含量,将有助于提高活性炭的产量及性能。

表1 单位质量样品的元素分析数据
从脱灰前后煤样品的热失重曲线 (图3)可看到,煤-D失重曲线比煤-R更陡峭。煤的热解反应非常复杂,生成气体(CO、H2、CH4、C2H4、CO2、C2H6等)、焦油和固定碳[18]。煤-R和煤-D热失重均分为4个阶段。煤-R的第一阶段从初温到200℃,曲线缓慢下降,主要为水分和吸附气体的挥发;第二阶段为210298℃,煤中弱键官能团发生分解,析出CO2为主的气体产物;第三阶段为298777℃,失重曲线急剧下降,煤热解释放出大量挥发分后,变成半焦,同时会进一步与空气中氧气发生氧化反应,分子链断裂成小分子气体,造成显著失重;第四阶段为777℃以后,失重曲线趋于平稳,所剩组成大多以灰分为主。对于煤-D,第一阶段从初温到200℃,第二阶段216286℃,第三阶段286776℃,第四阶段776℃以后。煤-R与煤-D最终质量残余率分别为19%和5%,表明脱灰后煤中不能热解的灰分含量显著降低。这与元素分析结果 (表1)所证实的脱灰有助于提高煤中碳元素含量是一致的。经比较,可以发现该煤-R的最终质量残余率小于通过工业分析所测得的灰分(27.29%),这是因为热重分析中所用的样品量较少,且升温速率小,使空气中的氧气能够充分扩散到煤样品深处,导致失重反应更加彻底。因灰分无助于活性炭的比表面积与孔隙率,所以本文所制备活性炭均经脱灰处理。

图3 脱灰对煤热失重曲线的影响
2.2 活化条件对活性炭结构与性能的影响
从XRD谱图(图4)可见,AC-Zn在26.58°处出现Zn2+的衍射峰。AC-K与AC-Zn在(002)面衍射角分别为22.7°和24.8°,由Bragg公式可知AC-Zn的碳层面间距较小、石墨化程度高。这是因为由ZnCl2活化得到活性炭的孔隙结构不稳定,易于石墨化[23]。因此,为防止过渡活化并石墨化而破坏AC的孔结构,应适当减少活化剂ZnCl2用量。

图4 活性炭的XRD谱图
在红外光谱(图5)可看到,AC-K与AC-Zn同时存在较强的C—O键(1102.84 cm-1)及—O—H键(29003600 cm-1)反射峰,且AC-Zn的C—O键(1102.84 cm-1)反射峰更尖锐。说明在活化过程中ZnCl2在活性炭表面产生大量对碳骨架具有保护作用的偏酸性官能团 (如C—O等),同时作为一种Lewis酸ZnCl2会促进芳烃聚合形成交联结构,而得到大量相关官能团[24]。
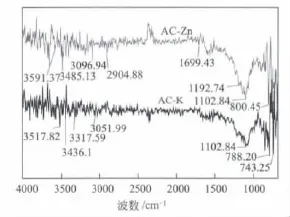
图5 活性炭的红外光谱图
表1给出的元素分析结果可见,AC-K与ACZn均不含N元素,且AC-Zn未测出H元素。一方面是因为ZnCl2的催化脱氢与脱水作用;另一方面是因KOH在活化过程中产生碱基自身脱水作用[25]。因为高温时K有极强供电子效应,易于与碳形成稳定的络合盐类,所以AC-K中C元素含量比AC-Zn高[26]。另外,KOH在活化过程中会生成活性氧物种,导致AC-Zn中O元素含量偏高。
随活化剂用量的提高,AC-K与AC-Zn碘值先增大后减小 (图6),分别在KOH/煤=3、ZnCl2/煤=2时达最高值。随KOH用量增加,会通过插层作用进到炭层间反应,生成大量微孔,增大碘值[27]。但当KOH量过高时,部分微孔扩大转变为中孔甚至大孔,导致碘值降低[19]。同理,ZnCl2为活化剂时也有类似趋势。此外,在相同活化温度与时间时,AC-Zn比AC-K碘值高,与XRD结果一致,是因其活化作用强所致。

图6 活化剂/煤配比及活化温度与碘值的关系
从图6也可看到,AC-K(KOH/煤=3)与AC-Zn(ZnCl2/煤=1)都在活化温度为700℃时碘值最低,800℃时碘值最高。当活化温度较低时,伴随羟基与水脱除的活化反应速率随温度升高而变快,导致微孔隙变多、碘值升高;当温度较高时,活化反应发展至难以快速脱离的聚合物或络合物,致使微孔减少、碘值降低;当进一步提高温度,上述较大分子又分解成易于挥发的小分子基团,形成丰富微孔隙、碘值增大。
2.3 活性炭吸附性能研究与应用
考虑到以KOH为活化剂制备的活性炭性能较差,且存在设备腐蚀问题。因此,从实用角度出发,本文重点研究了AC-Zn的吸附性能。首先,通过对有机蒸气的吸附量,计算得到活性炭的孔体积,与市售活性炭对比,可知本文所制AC-Zn的孔体积高4倍以上(表2)。说明本文AC-Zn具有更高的孔隙率和更好的应用前景。

表2 活性炭AC-Zn气相吸附数据(26℃,P/P 0=0.95)
当活性炭吸附时间为20 min时,随AC-Zn用量增加甲基橙的脱除率显著提高(图7),当AC-Zn用量小于0.8 g时,脱除率急速增加,而高于0.8 g后,增加幅度变缓,趋于平衡值85%。综合考虑到操作费用(活性炭用量)与脱除效果,优选1.0 g。此时,AC-Zn吸附效果显著高于市售活性炭的70%[19]。随吸附时间从5 min延至40 min,甲基橙的脱除率刚开始陡增(图7),当时间大于15 min,因吸附-解吸附速率趋于平衡,脱除率增加趋缓。
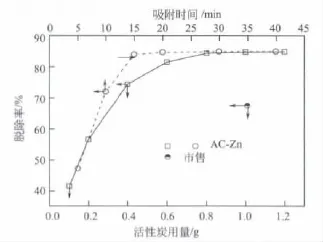
图7 活性炭用量及吸附时间对吸附效果的影响
由图8可知,随吸附温度从30℃提高至50℃时,甲基橙脱除率呈线性地迅速提高。温度升高对活性炭吸附具有明显促进作用,可能是由于温度提升,促进甲基橙的分子热运动,增加了与活性炭的碰撞概率,吸附提高。而从50℃再提高至60℃时,因高温有助于甲基橙从AC表面脱附,使吸附与脱附两过程趋于平衡,因而脱除率仅从84.8%略微提高到85.1%。随甲基橙溶液浓度从20 mg/L提高到120 mg/L时,脱除率几乎线性降低,说明随甲基橙量的提高,活性炭无法提供足够甲基橙吸附所需吸附表面所致。
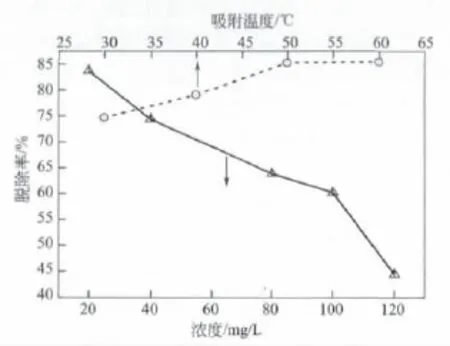
图8 温度及甲基橙浓度对吸附效果的影响
综合以上数据,可得到活性炭AC-Zn吸附脱除甲基橙的最佳条件为活性炭用量1 g,吸附时间15 min,温度50℃,脱除率高于85%。
3 结 论
以阜新烟煤经脱灰及活化等步骤成功制得较高性能活性炭。当以KOH为活化剂时,最佳条件为:碱煤比3,活化温度800℃,活化时间90 min;当以ZnCl2为活化剂时,最佳条件为:ZnCl2/煤比2,活化温度600℃,活化时间50 min。以ZnCl2作活化剂更有利于提高活性炭的吸附性能,其孔容积及吸附效果显著高于工业活性炭。随甲基橙水溶液的温度越高,活性炭用量越多,时间越长,吸附效果越好。甲基橙最佳吸附操作条件:活性炭用量1 g,吸附温度50℃,吸附时间15 min。
[1]Allen S J,Whitten L.The production and characterisation of activated carbons:A review[J].Dev.Chem.Eng.Mineral.Process,1998,6(5):231-261.
[2] 窦智峰,姚伯元.高性能活性炭制备技术新进展 [J].海南大学学报:自然科学版,2006,24(1):74-82.
[3]Maldhure A V,Ekhe J D.Preparation and characterizations of microwave assisted activated carbons from industrial waste lignin for Cu(II)sorption[J].Chemical Engineering Journal,2011,168(3):1103-1111.
[4]Yagmur E,Ozmak M,Aktas Z.A novel method for production of activated carbon from waste tea by chemical activation with microwave energy[J].Fuel,2008,87:3278-3285.
[5]Nabais J M V.Laginhasa C E C,Carrotta P J M,et al.Production of activated carbons from almond shell[J].Fuel Processing Technology,2011,92(3):234-240.
[6]Daud W M A W,Ali W S W,Sulaiman M Z.The effects of carbonization temperature on pore development in palm-shellbased activated carbon[J].Carbon,2000,38(14):1925-1932.
[7]Liou T H.Development of mesoporous structure and high adsorption capacity of biomass-based activated carbon by phosphoric acid and zinc chloride activation[J].Chemical Engineering Journal,2010,158:129-142.
[8]Qada E N E,Allen S J,Walker G M.Influence of preparation conditions on the characteristics of activated carbons produced in laboratory and pilot scale systems[J].Chemical Engineering Journal,2008,142(1):1-13.
[9]Kopac T.Preparation of activated carbons from Zonguldak region coals by physical and chemical activations for hydrogen sorption[J].International Journal of Hydrogen Energy,2007,32:5005-5014.
[10]Usmani T H.Preparation and characterization of activated carbon from a low rank coal[J].Carbon,1996,34(1):77-82.
[11]Hsu L Y,Teng H.Influence of different chemical reagents on the preparation of activated carbons from bituminous coal[J].Fuel Processing Technology,2000,64:155-166.
[13] 解强,乐政.含钾化合物在煤基活性炭制备中的作用 [J].中国矿业大学学报,1997,26(4):71-73.
[14] 乐政.化学添加剂在制备煤基活性炭中的作用与比较 [J].煤化工,1999,87(2):37-40.
[15] 邢雯雯,周铁桥,张军,等.煤基磁性活性炭的制备 [J].北京科技大学学报,2009,31(1):83-87.
[16] 邢宝林,谌伦建,张传祥,等.NaOH活化法制备煤基活性炭的研究[J].煤炭转化,2010,33(1):69-73.
[17] 邢宝林,张传祥,潘兰英,等.高比表面积煤基活性炭的制备及其吸附性能的研究[J].洁净煤技术,2008,14(1):85-88.
[18] 邢宝林,张传祥,谌伦建,等.配煤对煤基活性炭孔径分布影响的研究[J].煤炭转化,2011,34(1):43-46.
[19] 张传祥,张睿,成果,等.协同活化对煤基活性炭物化性能的调控[J].煤炭转化,2008,31(2):52-60.
[20] 中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.GB/T 212—2008,煤的工业分析方法[S].北京:中国标准出版社,2009.
[21]国家质量技术监督局.GB/T 12496.8—1999 B73,木质活性炭试验方法-碘吸附值的测定 [S].北京:中国标准出版社,2000.
[22]国家质量技术监督局.GB/T 12496.5—1999 B73,木质活性炭试验方法-四氯化碳吸附率 (活性)的测定 [S].北京:中国标准出版社,2000.
[23]谢招娣,马承愚,宋新山.KOH和ZnCl2活化剂对苎麻秆基活性炭微观结构的影响 [J].高分子材料科学与工程,2012,28(2):106-109.
[24]Ahmadpour A,Do D D.The preparation of activated carbon from macadamia nut shell by chemical activation[J].Carbon,1997,35(12):1723-1732.
[25] 张会平,叶李艺,杨立春.氯化锌活化法制备木质活性炭研究[J].材料科学与工艺,2006,14(1):42-45.
[26] 邢伟,张明杰,阎子峰.超级活性炭的合成及活化反应机理[J].物理化学学报,2002,18(4):340-345.
[27] 王秀芳.高比表面积活性炭的制备、表征及应用 [D].广州:华南理工大学,2006.
