当归中阿魏酸含量测定及质量标准的研究
2012-12-23王中华
王中华
(天津中医药大学第一附属医院药剂部,天津 300193)
制剂与质量控制
当归中阿魏酸含量测定及质量标准的研究
王中华
(天津中医药大学第一附属医院药剂部,天津 300193)
目的 参照高效液相色谱法(《中华人民共和国药典2010年版》一部附录ⅥD)试验,对当归中的主要成分阿魏酸进行含量测定。方法 根据不同的流动相、提取方法、提取时间、取样量、提取溶剂、浓缩温度,最终确定最佳的测定方法,即以乙腈—0.1%磷酸(22∶78)为流动相;检测波长为324 nm,取样量为4 g,70%甲醇回流提取30 min,80℃的水浴加热的浓缩温度。结果 根据实验并通过线性关系考察,证明线性关系良好。重复性、重现性、稳定性、回收率均符合要求。结论 此方法当归以阿魏酸(C10H10O4)计,不少于0.10 mg,可作为以当归为主要成分的中药制剂的质量检测标准和参考依据。
阿魏酸;当归;分析;色谱法,高压液相
当归是常用中药材之一,也是许多中成药组方中的常用品种,具有补血活血、调经止痛、润肠通便等功效。其主要成分有挥发油类、香豆素类、有机酸类、糖类、黄酮类、氨基酸、维生素和其他微量元素具有很高的药用价值。其中有机酸类中主要成分阿魏酸,具有抗氧化和清除自由基的作用,能有效抑制血小板聚集和血栓形成,可用于治疗多种心脑血管性疾病。我们利用高效液相色谱(HPLC)法测定当归的主要有效成分阿魏酸的含量,能较好地反映其相关成品的质量和疗效[1],可作为以当归为主要成分的中药制剂的质量检测标准。故参照《中华人民共和国药典2010年版》一部项下相关方法,制订了其HPLC法的含量测定标准和方法[2]。
1 HPLC法测定当归中阿魏酸
1.1 仪器与试药 仪器:LC-10ATVP岛津高效液相色谱仪;SPD—10A VP N2000数据工作站(浙江大学智能信息研究所);HP1100 DAD检测器;FA1104电子分析天平(上海精科天平厂);DK98-1型电热恒温水浴锅(双列六孔,天津中环科技开发公司);ACO-3120型真空泵(上海会行农机厂);超声处理器(北京医疗设备二厂);旋转蒸发仪(上海亚荣仪器公司);药典筛(浙江五星冲压筛具厂);101 2BS电热恒温干燥箱(天津中环实验电炉有限公司);ZDHW调温型电热套(5 000 mL,河北省黄骅中兴仪器有限公司);DL-1单联电炉(2 000 W,北京中兴伟业仪器有限公司);玻璃仪器等。色谱柱:Irregular-H C18,10 μm,250 mm ×4.6 mm。试剂:乙腈、磷酸,为色谱纯,其余甲醇等试剂均为分析纯;水为蒸馏水(自制)。对照品:阿魏酸(中国药品生物制品检定所,批号:110773-200911)。样品:黄地散(天津中医药大学第一附属医院制剂室提供,主要成分:当归、生地黄、何首乌、黄精等,批号:090426,090428,090429)。
1.2 方法学的考察
1.2.1 测定波长的选择与考察 《中华人民共和国药典2010年版》一部[2]当归项下,检测波长为316 nm,用岛津高效液相色谱仪,在波长200~600 nm进行扫描,阿魏酸在324 nm处有最大吸收峰,最终确定324 nm为检测波长。见图1。

图1 阿魏酸HPLC扫描图(200~600 nm)
1.2.2 流动相的选择与考察 本实验采用4个不同系统的流动相:乙腈-0.1%磷酸、乙腈-1%冰乙酸、甲醇-0.1%磷酸、甲醇-1%冰乙酸进行测定,同时考察了各系统不同组成比例的流动相测定[3]。
结果:流动相乙腈-0.1%磷酸(22∶78)阿魏酸出峰时间适中,约为14 min,分离度好,确定为最终的流动相。柱温:室温;流速:0.8 mL/min;理论板数按阿魏酸峰计算不低于3 000。
1.2.3 取样量的选择与考察 取同一批号(090426)样品,共 4 份,分别精密称定 2、4、6、8 g,置具塞锥形瓶中,精密加70%甲醇50 mL,密塞,称定质量,加热回流30 min,放冷,再称定质量,用70%甲醇补足减失的质量,摇匀,滤过,即得。在流动相乙腈-0.1%磷酸(22∶78)色谱条件下测定样品中阿魏酸峰面积。结果见图2~5和表1。

图2 样品(2 g)的阿魏酸HPLC图

图3 样品(4 g)的阿魏酸HPLC图

图4 样品(6 g)的阿魏酸HPLC图

图5 样品(8 g)的阿魏酸HPLC图

表1 样品不同取样量的阿魏酸含量测定结果 (n=2)
结果显示,取样量以2、4 g阿魏酸含量接近,但是2 g取样量所测得峰面积过小,不利于实验操作,故选择取样量为4 g。
1.2.4 提取方法的选择与考察 取同一批号(090426)样品,共2份,各4 g,分别精密称定,置具塞锥形瓶中,精密加70%甲醇50 mL,密塞,称定质量,其中1份加热回流30 min,另1份超声处理30 min,放冷,再称定质量,用70%甲醇补足减失的质量,摇匀,滤过,即得。在流动相乙腈-0.1%磷酸(22∶78)色谱条件下测定样品中阿魏酸峰面积,结果见图6、7和表2。

图6 超声提取的阿魏酸HPLC图

图7 回流提取的阿魏酸HPLC图
结果显示,回流提取测得样品的阿魏酸含量高于超声处理的样品,故确定回流的提取方法。
表2 不同提取方法的阿魏酸测定结果 (n=2)
1.2.5 提取溶剂的选择与考察 取同一批号(090426)样品,共2份,各4 g,分别精密称定,置具塞锥形瓶中,分别精密加70%甲醇、100%甲醇各50 mL,密塞,称定质量,加热回流30 min,放冷,再称定质量,分别各自用70%甲醇、100%甲醇补足减失的质量,摇匀,滤过,即得。在流动相乙腈-0.1%磷酸(22∶78)色谱条件下测定样品中阿魏酸峰面积,结果见图8、9和表3。

图8 100%甲醇提取的阿魏酸HPLC图

图9 70%甲醇提取的阿魏酸HPLC图

表3 不同溶剂提取的阿魏酸测定结果 (n=2)
结果显示,70%甲醇提取测得的阿魏酸含量高于100%甲醇提取的处理的样品,故确定70%甲醇为提取溶剂。
1.2.6 提取时间的选择与考察 取同一批号(090426)样品,共3份,各4 g,分别精密称定,置具塞锥形瓶中,分别精密加70%甲醇50 mL,密塞,称定质量,分别加热回流15、30、45 min,放冷,再称定质量,用70%甲醇补足减失的质量,摇匀,滤过,即得。在流动相乙腈 -0.1%磷酸(22∶78)色谱条件下测定样品中阿魏酸峰面积。结果见图10~12和表4。
图10 70%甲醇(15 min)提取的阿魏酸HPLC图
图11 70%甲醇(30 min)提取的阿魏酸HPLC图

图12 70%甲醇(45 min)提取的阿魏酸HPLC图

表4 不同回流提取时间的阿魏酸测定结果 (n=2)
结果显示,以回流提取30 min时,测定的阿魏酸含量为最高。
1.2.7 浓缩温度的选择与考察 为提高样品溶液的浓度,减少阿魏酸的加热损失,便于测定,考虑将样品溶液进行浓缩,本实验考察了60、80℃下的影响。取同一批号(090426)样品,共2份,各4 g,分别精密称定,置具塞锥形瓶中,分别精密加70%甲醇50 mL,密塞,称定质量,加热回流30 min,放冷,再称定质量,用70%甲醇补足减失的质量,摇匀,滤过,精密吸取续滤液25 mL,分别置60、80℃的水浴上浓缩至近干,残渣用70%甲醇溶解,转移至5 mL量瓶中,加70%甲醇至刻度,摇匀,滤过,即得。在流动相乙腈-0.1%磷酸(22∶78)色谱条件下测定样品中阿魏酸峰面积,结果见图13、14和表5。

图13 70%甲醇(60℃)提取的阿魏酸HPLC图

图14 70%甲醇(80℃)提取的阿魏酸HPLC图

表5 不同浓缩温度的阿魏酸测定结果
结果显示,虽然80℃的水浴加热浓缩温度高于60℃,容易造成阿魏酸的加热损失,但浓缩时间大大缩短,对阿魏酸的含量影响也不大,故选定80℃的水浴加热为浓缩的温度。
1.3 溶液的制备
1.3.1 对照品溶液的制备 取阿魏酸对照品适量,精密称定,加70%甲醇溶液制成每mL含8 μg的溶液,即得。
1.3.2 供试品溶液的制备 取装量差异项下的样品,研细,取约4 g,精密称定,置具塞锥形瓶中,精密加70%甲醇50 mL,密塞,称定质量,加热回流30 min,放冷,再称定质量,用70%甲醇补足减失的质量,摇匀,滤过,精密量取续滤液25 mL,置80℃水浴蒸至近干,残渣加70%甲醇适量溶解,移至5 mL量瓶中,加70%甲醇至刻度,摇匀,滤过,取续滤液,即得。

图15 阿魏酸对照品HPLC图
1.3.3 阴性对照液的制备 取缺少当归的处方药,按制备工艺制得样品,再按供试品溶液的制备方法,制得阴性样品溶液。进样后HPLC图中与阿魏酸相应位置上无吸收。见图15~17。

图16 阿魏酸供试品HPLC图

图17 阿魏酸阴性对照品HPLC图
1.4 重复性试验 取同一批号(090426)样品,取1份,研细,取4 g,按照“1.3.2”供试品溶液制备操作,按流动相乙腈-0.1%磷酸(22∶78)色谱条件分析,连续进样6次,测定样品中阿魏酸峰面积,测得峰面积值的RSD为1.11%,符合要求,结果见表6。

表6 重复性试验结果
1.5 重复性试验 取同一批号(090426)样品,共6份,研细,取4 g,按照“1.3.2”供试品溶液制备操作,按流动相乙腈-0.1%磷酸(22∶78)色谱条件分析,测定每份样品中阿魏酸含量。结果样品中阿魏酸平均含量为0.023 21 mg/g,RSD为1.32%,符合要求,结果见表7。

表7 重复性试验结果
1.6 稳定性试验 取同一批号(090426)样品,取1份,研细,取4 g,按照“1.3.2”供试品溶液制备操作,按流动相乙腈 -0.1%磷酸(22∶78)色谱条件分析,分别在 0、1、2、4、8、24 h,测定样品中阿魏酸峰面积,测得峰面积值的RSD为2.06%,符合要求,结果见表8。

表8 稳定性试验结果
1.7 线性关系的考察 对照品溶液的制备:取阿魏酸对照品适量,精密称定,加70%甲醇制成每mL含0.208 mg的溶液置作为储备液,再分别精密量取 0.2、1、2、4、6、8 mL,置25 mL量瓶中,加70%甲醇稀释至刻度,分别精密吸取上述6种浓度的对照品溶液各10 μL,注入高效液相色谱仪,按流动相乙腈-0.1%磷酸(22∶78)色谱条件分析,测定各自峰面积,以峰面积为横坐标,阿魏酸对照品的含量为纵坐标,绘制标准曲线。计算回归方程为:Y=3.09E -005X,r=1.000 0,表明阿魏酸在 0.016 64 ~0.665 6 μg范围内与峰面积线性关系良好,见表9、图18。

表9 线性关系考察测定结果

图18 阿魏酸对照品标准曲线图
1.8 加样回收率试验 取同一批号(090426)样品,共6份,研细,取2 g,精密称定,分别加入阿魏酸对照品溶液0.049 92 mg/mL各 1 mL,精密加入 70% 甲醇 49 mL,再按照“1.3.2”供试品溶液制备操作,制得供回收率用供试品溶液,按流动相乙腈-0.1%磷酸(22∶78)色谱条件分析,计算回收率,结果平均回收率为 101.76%,RSD为1.28%。结果见表10。

表10 加样回收率试验测定结果
2 样品中阿魏酸质量标准的测定
样品的制备:取装量差异项下的本品,研细,取约4 g,精密称定,置具塞锥形瓶中,精密加70%甲醇50 mL,密塞,称定质量,加热回流30 min,放冷,再称定质量,用70%甲醇补足减失的质量,摇匀,滤过,精密量取续滤液25 mL,置80℃水浴蒸至近干,残渣加70%甲醇适量溶解,移至5 mL量瓶中,加70%甲醇至刻度,摇匀,滤过,取续滤液即得。测定3批样品中阿魏酸含量。见表11。
由于当归中阿魏酸含量较低为0.05%,通过测定3批中试成品的含量,同时考虑使用药材的质量差异,按其转移率40%,作为含量限度。转移率是根据当归的投料量以及《中华人民共和国药典2010年版》中所规定的当归中阿魏酸的含量要求,算出总的阿魏酸含量,乘以40%,得出成品中要求的阿魏酸的最低含量,即每袋含阿魏酸不低于0.10 mg。
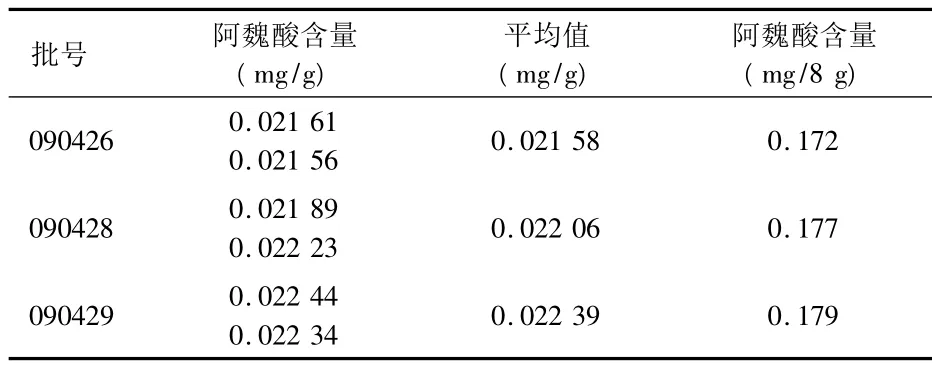
表11 样品含量测定结果 (n=2)
3 结论
以当归中的阿魏酸为含量指标,应用正交实验表进行实验分析,优选了最佳提取制备工艺,即煎煮3次,每次1.5 h,加10倍量水。结果表明制备工艺方法简单可靠,具有科学性和可行性。
质量标准:照高效液相色谱法(《中华人民共和国药典2010年版》一部附录VI D)[2]试验,对当归中的主要成分阿魏酸进行含量测定,根据不同的流动相、提取方法、提取时间、取样量、提取溶剂及浓缩温度,最终确定最佳的测定方案,即以乙腈-0.1%磷酸(22∶78)为流动相;检测波长为324 nm,理论板数按阿魏酸峰计算应不低于3 000。取样量为4 g,70%甲醇回流提取30 min,80℃的水浴加热的浓缩温度。并通过线性关系考察,证明线性关系良好。重复性、重现性、稳定性、回收率均符合要求。8 g/袋包装中,当归以阿魏酸(C10H10O4)计,不少于0.10 mg。可作为以当归为主要成分的中药制剂的质量检测标准和参考依据。
本实验提取工艺的确定应以制剂的安全、有效、稳定为最终目标。当归中阿魏酸在提取时会因受热而出现不稳定(损失)的现象,故应严格控制提取时的受热时间和温度。
本实验的质量标准主要是检测阿魏酸的含量,由于其受热后不稳定,而且转移率较低,对生产和鉴别会产生影响,未来还应设法提高阿魏酸的转移率和最低含量。
在方法学考察中发现,相关项目的色谱图的对比差别性不高。目标峰的峰面积较小,未来还应考虑对各项色谱条件进一步优化和提高。
[1] 王宝琴.中成药质量标准与标准物研究[M].北京:中国医药科技出版社,1994:309,615.
[2] 国家药典委员会.中华人民共和国药典2010年版:一部[M].北京:化学工业出版社,2010:488-489.
[3] 唐力英,王祝举,赫炎,等.高效液相色谱法测定当归中阿魏酸的含量[J].中国实验方剂学杂志,2006,12(2):14 -15.
Determination of ferulic acid in Angelica and research of quality standards
WANG Zhonghua.Department of Pharmacy,First Affiliated Hospital of Tianjin University of Traditional Chinese Medicine,Tianjin 300193
Objective To refer to high-performance liquid chromatography(《Chinese Pharmacopoeia》2010 of one appendix Ⅵ D)trial and determine the ferulic acid in the main components of Chinese angelica.Methods With different the mobile phase,extraction method,extraction time,sample volume,extraction solvent,concentration temperature,ultimately the best method for the determination was determined that Acetonitrile - 0.1%phosphoric acid(22 ∶78)as mobile phase;detection wavelength is 324 nm,sample volume is 4 g,70%methanol extraction and 30 min,the temperature of concentrating is 80 ℃ water bath heating.Results Through investigation proves the method of the linear relations is best.Repeatability,reproducibility,stability,recovery rates are to meet requirements.Conclusion Angelica calculated in accordance with ferulic acid(C10H10O4)is not less than 0.10 mg/8 g and this method can be as quality inspection standards and reference of a main component for angelica of traditional Chinese medicine preparation.
Ferulic acid;Angelica;Content determination;High-performance liquid chromatography
R282.71;R284.1;R932.41
A
1002-2619(2012)07-1058-06
王中华(1970—),男,主管药师,硕士。研究方向:中药制剂和化学分析。
2010-01-30)
