N-甲基吡咯-2-甲醛激发态结构动力学及其溶剂效应的共振拉曼光谱和密度泛函理论研究
2012-12-21许宗平赵彦英王惠钢郑旭明
许宗平 赵彦英 王惠钢 郑旭明
(浙江理工大学化学系,先进纺织材料与加工技术教育部重点实验室,生态染整技术教育部工程研究中心,杭州310018)
N-甲基吡咯-2-甲醛激发态结构动力学及其溶剂效应的共振拉曼光谱和密度泛函理论研究
许宗平 赵彦英 王惠钢 郑旭明*
(浙江理工大学化学系,先进纺织材料与加工技术教育部重点实验室,生态染整技术教育部工程研究中心,杭州310018)
获取了覆盖N-甲基吡咯-2-甲醛(NMPCA)A-带和B-带电子吸收共7个激发波长的共振拉曼光谱,并结合含时密度泛函理论(TD-DFT)方法研究了的A-带和B-带电子激发和Franck-Condon区域结构动力学. TD-B3LYP/6-311++G(d,p)计算表明:A-带和B-带电子吸收的跃迁主体为π→π*.共振拉曼光谱可以指认为, 11-13振动模式(A-带激发)或者7-11振动模式(B-带激发)的基频、倍频和组合频,其中C=O伸缩振动(ν7)、环的变形振动+N1-C6伸缩振动(ν17)、环的变形振动(ν21)和C6-N1-C2/C2-C3-C4不对称伸缩振动(ν14)占据了绝大部分.这表明NMPCA的Sπ激发态结构动力学主要沿C=O伸缩振动、环的变形振动和环上N1-C6伸缩振动等反应坐标展开.在同一溶剂的共振拉曼光谱中随激发波长由长变短,ν7与ν14的强度比呈现出由强变弱再变强的现象,这种变化规律被认为与Franck-Condon区域Sn/Sπ态混合或势能面交叉有关.溶剂对Sn/Sπ态混合或势能面交叉具有调控作用.
N-甲基吡咯-2-甲醛;激发态结构动力学;共振拉曼光谱;溶剂效应
1 引言
芳香羰基化合物的光诱导电子转移、氢转移和能量转移反应在化学、生物和材料科学等领域起着很重要的作用.近几十年来,人们对以苯甲醛和苯乙酮为代表的芳香羰基化合物的光诱导系间窜跃、磷光、异构化、键断裂和原子转移等光物理和光化学过程开展了大量研究.
由于环与羰基之间的共轭作用,芳香羰基化合物的电子激发态能量和反应性能与脂肪族羰基化合物显著不同.在光化学反应方面,实验研究发现,处于1nπ*和3nπ*电子态的脂肪酮类化合物都可以发生NorrishI和NorrishII反应,但大多数的芳香醛酮化合物却只能在最低三态上发生上述反应.1-3苯甲醛和苯乙酮经紫外激发后都发生光解离反应,但苯甲醛主要生成苯和一氧化碳,4-6而苯乙酮生成苯甲酰基和甲基自由基.7-10在光物理方面,芳香醛酮化合物具有很强的磷光,但是荧光很弱,其单重激发态寿命比相应的脂肪酮寿命要小的多.苯甲醛和苯乙酮的磷光光谱研究表明,由光激发到1nπ*态引发的高磷光产率被证明与非常有效的1nπ*(S1)-3ππ*(暗态T2)耦合11-16和T2(3ππ*)-T1(3nπ*)态混合6,16-18直接相关.方维海等19-21运用完全活性空间自洽场(CASSCF)方法对一系列芳香羰基化合物的几个最低电子态及其极小能量交叉点结构进行研究,发现S1/T2/T1三面交叉点,预示了芳香羰基化合物的暗态(S1,T2,T1)的结构、光物理和光反应性质.最近,Zewail等22-24用超快电子衍射技术研究了3nπ*态(暗态)的结构,获得与理论预示相一致的实验研究结果.
与S1(1nπ*)为暗态不同,芳香羰基化合物S2(1ππ*)为明态,光吸收效率高.但其激发态动力学和弛豫机制的研究还不多见.苯乙酮S2(1ππ*)激发态动力学的共振拉曼(FT-Raman)光谱学研究已有报道,25对S2态结构动力学和S2-S1耦合机制作了初步研究.因此,本文采用共振拉曼光谱学技术,结合量子化学计算,研究N-甲基吡咯-2-甲醛(NMPCA)S3、S2(1ππ*)态的结构动力学,考察杂环芳香醛S2(1ππ*)-S1(1nπ*)
耦合机制及其溶剂效应.
2 实验部分
2.1 试剂和仪器
NMPCA(98%,东京化成贩壳株式会社公司,日本);环己烷(光谱纯,99.5%,TEDIA公司,美国);乙腈试剂(光谱纯,99.9%,安徽时联特种试剂股份有限公司).
傅里叶变换拉曼光谱仪(Thermo Nicolet FT Raman 960 Spectrometer,美国);傅里叶变换红外(FTIR)光谱仪(Thermo-Nicolet FTIR Avatar 370,美国);共振拉曼光谱仪(自制).
2.2 实验方法
分别用环己烷、乙腈和水做溶剂,NMPCA的浓度约为4.00×10-3mol·L-1.共振拉曼光谱实验装置和方法参照文献.26由Nd:YAG纳秒脉冲激光器产生532.0、354.7和266.0 nm激光,它们经氢气受激拉曼位移管产生223.1、245.9、252.7、273.9、282.4和299.1 nm激光.采用流动循环方式进样.拉曼散射信号采用背向散射几何结构,经椭球镜等聚焦于单色仪的入口狭缝,由光栅分光后进入液氮冷却的电荷-耦合检测器(CCD)搜集.信号经25-35次累加,最后得到共振拉曼光谱.共振拉曼光谱的振动频率通过与环己烷拉曼谱带的已知频率值比较进行校正.从样品溶液的共振拉曼光谱减去纯环己烷、乙腈和水溶剂的拉曼光谱,得到7个激发波长下NMPCA分别在环己烷、乙腈和水溶剂中的共振拉曼光谱.其中,共振拉曼光谱的自吸收校正原理和实验装置中信号收集系统的强度校正原理参见文献,27具体校正通过Origin软件的自编程序完成.
3 理论计算
电子基态几何结构优化和简正振动频率在B3LYP/6-311++G(d,p)水平上计算获得,NMPCA分子为Cs点群.电子跃迁能在TD-B3LYP/6-311++ G(d,p)水平上计算得到.在计算复合物的结合能时,对自由基函数引起的基组叠加误差(BSSE)采用Boys和Bernardard的完全均衡校正法加以消除.校正后相互作用能的计算公式如下:

图1 NMPCA的顺式和反式构象异构体的结构示意图Fig.1 Schematic diagrams of cis and trans geometry structures of NMPCA
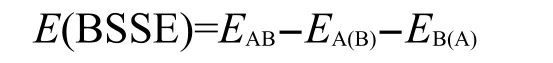
其中EAB是复合物AB的能量,EA(B)是复合物中B所有原子核设为携带虚轨道的傀儡原子时计算获得的单体A的能量,EB(A)是复合物中A所有原子核设为携带虚轨道的傀儡原子时计算得到的单体B的能量.本文所有计算均由Gaussian 03W程序包28完成.
4 结果和讨论
4.1 基态几何结构
为确定基电子态时NMPCA的能量极小点结构,对图1所示的四种可能的构象异构体进行了几何结构优化和振动频率计算.结果表明顺式构象异构体b和反式构象异构体c各有一个虚频,说明它们在B3LYP/6-311++G(d,p)势能面上为鞍点结构.顺式构象异构体a和反式构象异构体d为能量极小点结构,而顺式构象异构体a在能量上较反式构象异构体d低14.70 kJ·mol-1.
4.2 振动光谱
目前,对NMPCA的结构、振动光谱的实验测定和光谱指认及相关的理论计算等尚无文献报道.为了开展共振拉曼光谱指认和激发态结构动力学研究,本文计算了构象异构体a和d的拉曼光谱,测定了FT-IR和FT-Raman光谱(见补充材料图S1和S2, www.whxb.pku.edu.cn).结果表明,构象异构体a的计算与实验拉曼光谱最吻合,说明在常温下NMPCA主要以顺式构象异构体a的结构形式存在.表1列出了FT-IR和FT-Raman光谱的振动频率、顺式构象a的计算频率和光谱指认.
图2为NMPCA在环己烷、乙腈和水溶剂中的FT-Raman光谱.由图2可见,随着溶剂介电常数的增大(环己烷2.02,乙腈36.64,水78.39),νC=O谱带向低波数方向移动.与在环己烷溶液中νC=O谱带位于1672 cm-1相比,在乙腈溶液中NMPCA的νC=O谱带位于1666 cm-1,降低了6 cm-1,而在水溶液中位于1641 cm-1,降低了31 cm-1.其他振动模受溶剂的影响不明显.这说明溶剂极性和氢键对C=O伸缩振动频率产生了重要影响.
为了探讨氢键对C=O伸缩振动频率位移的影响,采用CPCM溶剂模型在B3LYP/6-311++G(d,p)计算水平上对氢键簇合物NMPCA-nH2O(n=1,2)的可能结构进行了结构优化,最终确定了6个能量极小点构象异构体,获得了它们的计算振动频率.图3示出氢键簇合物能量极小点构象异构体的结构示意图.体现氢键簇合物取向和氢键对C=O键长影响的部分结构参数示于其中.由图3可见,NMPCA的氧原子是质子受体,水中的氢原子是质子供体. NMPCA与水分子形成1:1和1:2氢键簇合物分别使C=O键长(CPCM水溶剂模型0.1232 nm)增长约0.0004-0.0006 nm和0.0005-0.0011 nm.表2列出了经过基组重叠误差校正后氢键簇合物6个能量极小点构象异构体的结合能和νC=O的振动频率(位移).由于在CPCM等溶剂模型下Gaussian 03软件不能计算基组重叠误差(BSSE),结合能(EB)数据是在气相条件下计算得到的.表2和表1说明,考虑水的溶剂化效应时,C=O的计算频率从气相时的1724 cm-1(表1ν7)降到1660 cm-1,降低了64 cm-1,这表明偶极耦合相互作用是导致羰基振动频率向低波数位移的一个主要原因.进一步考虑水簇合物时,氢键取向和数目会影响νC=O的计算频率值.取向不同时,氢键的键长不同,C=O键伸长的程度也不同.C=O键伸长越大,νC=O计算频率值越小.增加氢键的数目有利于降低氢键簇合物的能量和νC=O计算频率值.例如,表2和图3中NMPCA-2H2O cluster 4的结合能的能量较NMPCA-H2O cluster 2低27.25 kJ· mol-1,计算振动频率进一步降低9 cm-1.这说明氢键相互作用是羰基振动频率向低波数位移的另一个主要原因.图2显示,在水溶液中NMPCA的羰基振动谱峰(1641 cm-1)明显变宽,这是NMPCA与水的氢键相互作用的一个重要光谱佐证.

图2 NMPCA在纯液体、环己烷、乙腈和水中的FT-Raman光谱Fig.2 FT-Raman spectra of NMPCAin pure liquid and solvents of cyclohexane,acetonitrile,and water

表1 NMPCA理论振动频率和实验谱带的指认Table 1 Assignment of theoretical vibrational frequencies to experimental bands of NMPCA

表2 由B3LYP/6-311++G(d,p)和CPCM溶剂模型计算获得的水溶剂中NMPCA-nH2O(n=1,2)簇合物C=O伸缩振动的计算拉曼频率(νC=O)和由B3LYP/6-311++G(d,p)计算获得的气相条件下NMPCA-nH2O(n=1,2)簇合物的稳定能(Es)和结合能(EB)Table 2 Calculated Raman frequencies(νC=O)for C=O stretch of NMPCA-nH2O(n=1,2)clusters in water solvent,and the stabilization energy(Es)and binding energy(EB)of NMPCA-nH2O(n=1,2)clusters in gas phase using B3LYP/6-311++G(d,p) level of theory and/or employing CPCM solvent model
4.3 电子光谱
图4示出NMPCA在环己烷、乙腈和水溶剂中的紫外吸收光谱.223.1、245.9、252.7、266.0、273.9、282.4和299.1 nm为共振拉曼实验所用的激光波长.图4可见,不同溶剂对紫外吸收光谱的摩尔消光系数影响不大,但对最大吸收波长的位移影响明显.在上述3种溶剂中,NMPCA的最大吸收波长分别位于约280、285和293 nm处.随着溶剂极性增大,最大吸收波长λmax向低波数方向移动.表3列出在TDB3LYP/6-311++G(d,p)计算水平下NMPCA在气相时的电子跃迁能(ΔE)和振子强度(f).由于溶质分子在环己烷等非极性溶剂中的性质与气相条件下比较接近,表3选取了环己烷溶液中实验数据作为理论计算值的对比.由表3可见,计算给出2个允许电子跃迁带(259和245 nm),其f>0.1,与实验值280 nm(f=0.2622)和255 nm(f=0.2465)强吸收带基本一致.表3中,最高占有分子轨道(HOMO)和第三最高占有分子轨道(HOMO-2)是π成键轨道,分别表示为πH和πH-2.第二最高占有分子轨道(HOMO-1)是羰基氧上的非键轨道n.最低空价键轨道(LUMO)和第二最低空价键轨道(LUMO-1)是π*反键轨道和在和能隙之间,有三个弥散轨道.30第一弥散轨道(Ryd1)的密度涵盖-CH3的三个H原子和C5-H原子的外部区域,第二弥散轨道(Ryd2)的密度包含C3-H、C4-H、C7-H三个面内H原子和甲基面内H原子外部区域,而第三弥散轨道(Ryd3)的密度包含C4-H、C5-H、C7-H三个面内H原子和甲基面外两个H原子的外部区域.振子强度数据表明,在气相条件下,πH→π*L和πH-2→π*L跃迁是紫外吸收的主体.而暗态nH-1→π*L跃迁对总振子强度的贡献可以忽略,但可能在激发态势能面交叉方面发挥重要作用.表4列出NMPCA在CPCM溶剂模型下计算得到的电子跃迁能和振子强度.由表4可知,溶剂极性的增加使S1(1nπ*)暗态的能量升高,S2(1ππ*)和S3(1ππ*)明态的能量降低.氢键的作用使S1(1nπ*)暗态的能量进一步升高,S2(1ππ*)和S3(1ππ*)明态的能量进一步降低.这一预示与实验结果一致,即水溶剂使NMPCA紫外光谱的A-带和B-带的最大吸收波长发生了最大的红移,乙腈溶剂其次.

图3 NMPCA-nH2O簇合物的各种构型Fig.3 Schematic diagrams for different conformations of the NMPCA-nH2O clustersn=1,2;bond length in nm

图4 NMPCA在环己烷、乙腈和水溶剂中的紫外吸收光谱Fig.4 UV absorption spectra of NMPCAin cyclohexane, acetonitrile,and water solvents The excitation wavelengths used for the resonance Raman experiments are indicated above the absorption spectra.
4.4 共振拉曼光谱和激发态结构动力学
经过强度校正和溶剂扣减后的NMPCA在环己烷、乙腈和水中不同波长下的共振拉曼光谱见图5.图6为NMPCA在环己烷和水溶液中的282.4 nm共振拉曼光谱和它们的光谱指认.在图5和图6中,299.1、282.4和273.9 nm共振拉曼光谱的强度模式主要隐含了S2态激发态结构动力学信息(对应于A-带吸收),而252.7、245.9、223.1 nm共振拉曼光谱的强度模式主要隐含了S3态激发态结构动力学信息(对应于B-带吸收).B-带共振拉曼光谱可以被指认为7-11个Franck-Condon活性振动模的基频、倍频和它们的合频:ν7(C=O伸缩振动)、ν8(C2-C3-C4-C5不对称伸缩振动+C2-C7伸缩振动)、ν9(甲基剪式振动)、ν14(C6-N1-C2/C2-C3-C4不对称伸缩振动)、ν15(环的呼吸振动)、ν17(环的变形振动+N1-C6伸缩振动)、ν18(环上CH面内摇摆+甲基面内摇摆)、ν21(环变形振动)、ν22(环变形振动+C2-C7-C8面内摇摆)、ν23(N1-C6伸缩振动).
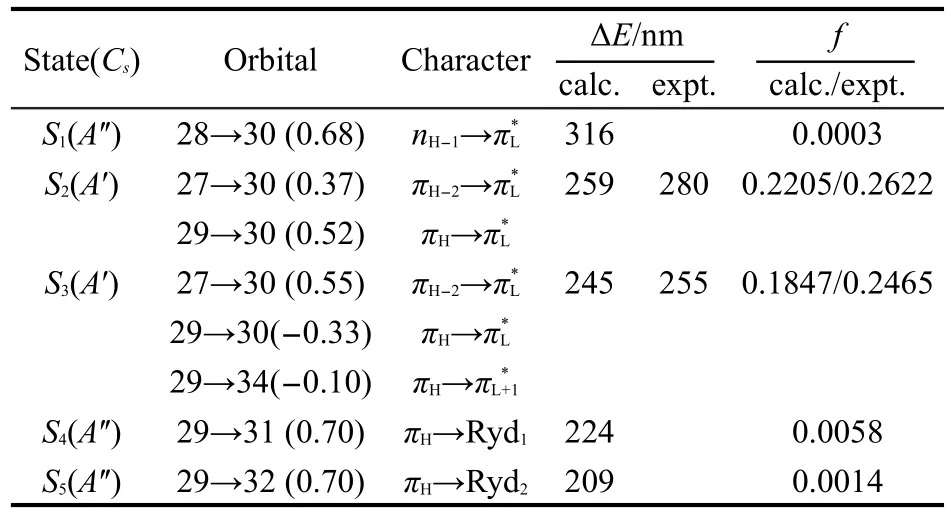
表3 气相中计算获得的NMPCA电子跃迁能(ΔE)和振子强度(f)Table 3 Calculated electronic transition energies(ΔE) and oscillator strengths(f)of NMPCAin gas phase
与B-带共振拉曼光谱的强度模式明显不同,A-带共振拉曼光谱的强度模式可以被指认为11-13个Franck-Condon活性振动模的基频、倍频和它们的组合频:ν7(C=O伸缩振动)、ν8(C2-C3-C4-C5不对称伸缩振动+C2-C7伸缩振动)、ν9(甲基剪式振动)、ν14(C6-N1-C2/C2-C3-C4不对称伸缩振动)、ν15(环的呼吸振动)、ν17(环的变形振动+N1-C6伸缩振动)、ν18(环上CH面内摇摆+甲基面内摇摆)、ν19(C3H9面内摇摆+甲基面内摇摆)、ν21(环变形振动)、ν22(环变形振动+C2-C7-C8面内摇摆)、ν23(N1-C6伸缩振动)、ν24(C3-C2-C7面内摇摆).ν8(1525 cm-1)和ν15(1308 cm-1)拉曼强度随激光波长从223.1 nm的中等强度到299.1 nm变得很弱,表明在A-带共振拉曼光谱中,这两个振动模的强度很可能来自B-带的预共振效应.31-35A-带共振拉曼光谱中,对激发态动态结构起决定作用的是ν7(C=O伸缩振动)、ν14(C6-N1-C2/ C2-C3-C4不对称伸缩振动)、ν17(环的变形振动+ N1-C6伸缩振动),它们的基频、泛频和合频占据了共振拉曼光谱强度的绝大部分,其次是ν18、ν21和ν23.这说明NMPCA的π→π*激发态结构动力学主要沿C=O伸缩振动、环的变形振动和N1-C6伸缩振动等反应坐标展开.

表4 NMPCA在CPCM溶剂模型下不同溶剂中的电子跃迁能(ΔE)和振子强度(f)Table 4 Calculated electronic transition energies(ΔE)and oscillator strengths(f)of NMPCAin different solvents using CPCM model

图5 NMPCA分别在环己烷(a)、乙腈(b)和水(c)中不同波长下的共振拉曼光谱Fig.5 Resonance Raman spectra of NMPCAin cyclohexane(a),acetonitrile(b),and water(c)solvents The spectra were obtained with the excitation wavelengths(in nm)indicated next to each spectrum.The spectra have been intensity-corrected and solvent-subtracted.Asterisks mark regions where solvent subtraction artifacts are present,and pound signs stand for residual uncertain laser.
面内环弯曲变形振动模ν21的拉曼强度与激发波长和溶剂有关.在环己烷和乙腈中ν21/ν14的相对强度变化不大,但在水中按273.9、282.4和299.1 nm激发光波长顺序依次增强.羰基伸缩振动模ν7的强度随激光波长呈现出有趣的变化.首先,在三种溶剂中按激光波长从长到短的顺序,该振动模的强度均呈现出由强变弱再变强的现象.在环己烷和乙腈中,最弱时出现在252.7 nm共振拉曼光谱中,而在水溶液中,最弱时出现在266.0 nm共振拉曼光谱中.其次,ν7/ν14强度比与溶剂的性质有很大关系,在(非质子性、非极性的)环己烷溶剂中最大,(强极性的)乙腈溶剂中居中,(质子性、强极性的)水溶剂中最小.羰基伸缩振动模ν7的这种强度变化规律可能直接体现了Franck-Condon区域Sn/Sπ态混合或激发态势能面交叉.这是因为,根据Franck-Condon原理和π、π*轨道特性,在没有Sn/Sπ态混合或势能面交叉的情况下,纯粹的π→π*跃迁不会在Franck-Condon区域产生重要的C=O键级变化,因而难以产生强烈的C=O伸缩振动强度.同样,纯粹的n→π*跃迁尽管在动力学上可以产生重大的C=O键级变化,但由于很弱的振子强度,也不会产生强烈的C=O伸缩振动强度.只有当发生Sn/Sπ态混合或势能面交叉时,根据振动-电子耦合原理,n→π*跃迁才可能从偶极允许的π→π*跃迁中借到强度,并在共振拉曼光谱中产生强度中等,甚至强度很大的C=O伸缩振动峰.根据这一分析,结合共振拉曼光谱中C=O伸缩振动峰的强度变化规律,我们认为在环己烷和乙腈溶剂中,最有效的Sn/Sπ态混合或势能面交叉发生在299.1 nm激发波长对应的能量附近,因为这时C=O伸缩振动具有最大的强度.水与NMPCA形成氢键簇合物不利于在Franck-Condon区域发生Sn/Sπ态混合或势能面交叉,因为在水溶液中C=O伸缩振动模的强度远小于其在环己烷和乙腈溶剂中的强度.一种合理的解释可能是由于水与C=O基形成氢键使n→π*跃迁的能量升高和Sn激发态势能面的显著位移,导致Sn/Sπ态混合或势能面交叉的位置离开了Franck-Condon区域,所以从共振拉曼光谱行为上表现为C=O伸缩振动模的强度减弱.这种溶剂对势能面交叉的影响最初由Improta等36提出,并用于解释核酸碱基Sn/Sπ势能面交叉的溶剂调控规律.

图6 NMPCA在282.4 nm环己烷(a)和水溶液(b)中的共振拉曼光谱指认Fig.6 Tentative vibrational assignments of the 282.4 nm resonance Raman spectra of NMPCAin cyclohexane(a)and water(b) The spectra have been intensity-corrected and solvent-subtracted.
5 结论
采用共振拉曼光谱技术和密度泛函理论方法研究了N-甲基吡咯-2-甲醛(NMPCA)的A-带和B-带电子激发和Franck-Condon区域结构动力学及其溶剂作用,得出如下结论:(1)NMPCA的A-带和B-带的跃迁主体都是π→π*的跃迁.C=O伸缩振动、环的变形振动和N1-C6伸缩振动的基频、泛频和合频占据了A-带和B-带共振拉曼光谱强度的绝大部分,表明Sπ激发态结构动力学主要沿这些反应坐标展开; (2)不同溶剂对实验拉曼光谱和紫外光谱具有明显的影响,溶剂主要与NMPCA分子中的羰基发生相互作用,氢键和偶极耦合相互作用是导致羰基振动频率向低波数位移的两个主要原因;(3)在同一溶剂的共振拉曼谱中,C=O伸缩振动模ν7随激发波长的强弱变化与Franck-Condon区域Sn/Sπ态混合或激发态势能面交叉强弱的变化有关.水与NMPCA形成氢键簇合物不利于Franck-Condon区域Sn/Sπ态混合或势能面交叉的发生.
(1) Wagner,P.J.Acc.Chem.Res.1971,4,168.
(2) Gilbert,A.;Baggott,J.Essentials of Molecular Photochemistry; CRC Press:Boca Raton,FL,1991.
(3)Horspool,W.;Armesto,D.Organic Photochemistry-A
Comprehensive Treatment;Ellis Horwood:New York,1992.
(4) Smolarek,J.;Zwarich,R.;Goodman,L.J.Mol.Spectrosc. 1972,43,416.
(5)Abe,H.;Kamei,S.;Mikami,N.;Ito,M.Chem.Phys.Lett.1984, 109,217.
(6) Ohmori,N.;Suzuki,T.;Ito,M.J.Phys.Chem.1988,92,1086.
(7) Robin,M.B.;Kuebler,N.A.J.Am.Chem.Soc.1975,97,4822.
(8) Berger,M.;Steel,C.J.Am.Chem.Soc.1975,97,4817.
(9) Zhao,H.Q.;Cheung,Y.S.;Liao,C.L.;Liao,C.X.;Ng,C.Y.; Li,W.K.J.Chem.Phys.1997,107,7230.
(10)Anand,S.;Zamari,M.M.;Menkir,G.;Levis,R.J.;Schlegel,H. B.J.Phys.Chem.A 2004,108,3162.
(11) Hirata,Y.;Lim,E.C.J.Chem.Phys.1980,72,5505.
(12) Hirata,Y.;Lim,E.C.Chem.Phys.Lett.1980,71,167.
(13) Koyanagi,M.;Goodman,L.;Chem.Phys.1979,39,237.
(14) Hayashi,H.;Nagakura,S.Mol.Phys.1974,27,969.
(15)Koyanagi,M.;Zwarich,R.J.;Goodman,L.J.Chem.Phys. 1972,56,3044.
(16) Kiritani,M.;Yoshii,T.;Hirota,N.;Baba,M.J.Phys.Chem. 1994,98,11265.
(17) Villa,E.;Amirav,A.;Chen,W.;Lim,E.C.Chem.Phys.Lett. 1988,147,43.
(18) Sneh,O.;Cheshnovsky,O.J.Phys.Chem.1991,95,7154.
(19)Fang,W.H.;Phillips,D.L.ChemPhysChem 2002,3,889.
(20)Wang,Y.W.;He,H.Y.;Fang,W.H.J.Mol.Struct.-Theochem 2003,634,281.
(21) Ding,W.J.;Fang,W.H.Progress in Chemistry 2007,19, 1449.[丁万见,方维海.化学进展,2007,19,1449.]
(22) Srinivasan,R.;Feenstra,J.S.;Park,S.T.;Xu,S.J.;Zewail,A. H.Science 2005,307,558.
(23) Feenstra,J.S.;Park,S.T.;Zewail,A.H.J.Chem.Phys.2005, 123,221104.
(24) Park,S.T.;Feenstra,J.S.;Zewail,A.H.J.Chem.Phys.2006, 124,174707.
(25) Ma,Y.;Pei,K.;Zheng,X.;Li,H.Chem.Phys.Lett.2007,449, 107.
(26)Li,S.P.;Wu,G.M.;Zheng,X.M.Chem.J.Chin.Univ.2004, 25,1495.[李少鹏,吴光明,郑旭明.高等学校化学学报, 2004,25,1495.]
(27) Myer,A.B.;Li,B.;Ci,X.J.Chem.Phys.1988,89,1876.
(28) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision B.02;Gaussian Inc.:Pittsburgh,PA,2003.
(29) Rauhut,G.;Pulay,P.J.Phys.Chem.1995,99,3093.
(30) Marian,C.M.J.Chem.Phys.2005,122,104314.
(31) Galica,G.E.;Johnson,B.R.;Kinsey,J.L.;Hale,M.O.J.Phys. Chem.1991,95,7994.
(32) Markel,F.;Myers,A.B.J.Chem.Phys.1993,98,21.
(33) Phillips,D.L.;Myers,A.B.J.Chem.Phys.1991,95,226.
(34)Kwok,W.M.;Phillips,D.L.J.Chem.Phys.1996,104,9816.
(35) Zheng,X.;Phillips,D.L.Chem.Phys.Lett.1998,286,79.
(36) Santoro,F.;Barone,V.;Gustavsson,T.;Improta,R.J.Am. Chem.Soc.2006,128,16312.
August 30,2011;Revised:October 15,2011;Published on Web:November 7,2011.
Resonance Raman Spectroscopy and Density Functional Theory Investigations on the Excited State Structural Dynamics of N-Methylpyrrole-2-carboxaldehyde and Its Solvent Effect
XU Zong-Ping ZHAO Yan-Ying WANG Hui-Gang ZHENG Xu-Ming*
(Key Laboratory of Advanced Textiles Materials and Manufacture Technology,and Engineering Research Center for Eco-dyeing and Finishing of Textiles,Ministry of Education,Department of Chemistry,Zhejiang Sci-Tech University,Hangzhou 310018,P.R.China)
Resonance Raman spectra of N-Methylpyrrole-2-carboxaldehyde(NMPCA)were obtained and seven excitations covered the A-and B-band electronic absorptions.The electronic excitations and the Franck-Condon region structural dynamics of NMPCA were studied by resonance Raman spectroscopy and time-dependent density functional theory(TD-DFT)calculations.The A-and B-band electronic absorptions were assigned to π→π*transitions on the basis of the TD-B3LYP/6-311++G(d,p)level of theory.The resonance Raman spectra showed Raman intensity in the fundamentals,the overtones and the combination bands for about 11-13 vibrational modes(A-band excitation)or 7-11 vibrational modes (B-band excitation).These were predominately due to the C=O stretch mode ν7,the ring deformation+N1-C6stretch ν17,the ring deformation mode ν21and the C6-N1-C2/C2-C3-C4anti-symmetry stretch mode ν14. This indicates that the Franck-Condon region Sπstructural dynamics of NMPCA mainly occurs along the C=O stretch,the ring deformation,and the N1-C6stretch reaction coordinates.In a certain solvent and under different excitation wavelengths the relative intensity of the C=O stretch mode ν7versus the C6-N1- C2/C2-C3-C4anti-symmetry stretch mode ν14shows an intense to weak to intense change as the excitation wavelengths decrease.This intensity variation directly reflects the Sn/Sπstate-mixing or crossing of the potential energy surfaces in the Franck-Condon region.Solvents can efficiently tune the Franck-Condon region Sn/Sπstate-mixing or crossing processes.
N-Methylpyrrole-2-carboxaldehyde;Excited state structural dynamics;Resonance Raman spectrum;Solvent effect
10.3866/PKU.WHXB20122865
*Corresponding author.Email:zxm@zstu.edu.cn;Tel:+86-571-86843699.
The project was supported by the National Key Basic Research Program of China(2007CB815203),National Natural Science Foundation of China (21033002,20803066),and Natural Science Foundation of Zhejiang Province,China(Y4090161).
国家重点基础研究发展规划(2007CB815203),国家自然科学基金(21033002,20803066)及浙江省自然科学基金(Y4090161)资助项目
O641;O643
