应用分布活化能模型分析伊敏褐煤丝炭腐植酸热解及氢气生成动力学
2012-12-21王传格曾凡桂彭志龙
王传格 曾凡桂 彭志龙 李 霞 张 莉
(太原理工大学煤科学与技术教育部及山西省重点实验室,太原030024;太原理工大学地球科学与工程系,太原030024)
应用分布活化能模型分析伊敏褐煤丝炭腐植酸热解及氢气生成动力学
王传格 曾凡桂*彭志龙 李 霞 张 莉
(太原理工大学煤科学与技术教育部及山西省重点实验室,太原030024;太原理工大学地球科学与工程系,太原030024)
应用开放体系的热重-质谱(TG-MS)联用技术在5、20和50°C·min-1三个升温速率下对伊敏褐煤丝炭腐植酸(F-HA)和脱灰丝炭腐植酸(DF-HA)的热解行为进行了分析,并利用分布活化能模型(DAEM)分别对其热解和氢气生成动力学进行分析,获得了热解过程和氢气生成的活化能分布函数.结果表明:(1)F-HA热解活化能分布函数呈类Gaussian分布,且具有一定的对称性,主峰位与标准Gaussian分布相一致;DF-HA热解活化能分布函数也呈类Gaussian分布形式,主峰位与Gaussian分布相比,偏向低活化能值.根据转化率与温度、活化能的关系,结合腐植酸的热失重特征,将F-HA热解过程划分为四个阶段,DF-HA热解过程划分为五个阶段,并对腐植酸热解过程中各阶段的化学反应进行了详细讨论.(2)F-HA和DF-HA热解氢气生成活化能分布函数均呈类Gaussian分布,热解氢气生成活化能的整体趋势为随着转化率的升高而增加,但也表现出一定的阶段集中分布特征.根据热解氢气生成的动力学特征,将其生成过程划分为五个阶段,反映了其生成的不同化学反应机制.(3)酸洗脱灰对伊敏丝炭提取出的HA的热解行为、热解过程及热解氢气生成动力学产生了影响.
腐植酸;热解;氢气生成;分布活化能模型;动力学;丝炭
1 引言
煤化作用是煤田地质学、有机地球化学等学科的核心内容之一,煤化作用的化学机制是煤中各种物质在热力作用下发生分解、聚合以及相互作用的总和.因此,系统地分析煤中各种物质在热力作用下发生的化学反应是深入认识煤化作用化学机制的基础.腐植酸(humic acid,简称HA)与有机溶剂可溶物、溶剂不可溶物一样,是褐煤的重要组成部分,是成煤过程中经过生物化学以及生物地球化学作用形成的以芳香结构为基本结构单元的无定形酸性物质.1现有的研究表明,在煤化作用过程中,腐植酸将进一步缩合,形成二氧化碳、水以及其它的烃类小分子物质及大分子物质.2虽然认识到了这种现象,由于腐植酸的组成及结构的复杂性,对其在煤化作用过程中的化学反应实质及过程的认识仍不清楚,这无疑将制约对煤化作用化学机制的认识.
近年来,随着氢洁净能源研究的兴起,煤制氢也成为煤转化研究的热点.氢气作为煤热解过程中的主要产物,一般认为是热解过程中缩聚反应的结果,但由于煤中氢赋存状态的复杂性,对其生成的动力学过程及机制仍未有系统的认识.氢气生成动力学的分析是认识氢生成反应类型的重要途径,这可以从煤热解过程中甲烷的生成反应动力学过程看出,如Cramer3认为煤热解甲烷的生成是多种反应共同作用的结果,每一种反应与其初始前驱物的化学活性位点有关,这些位置与一定的碳同位素信号相对应.他假定热解甲烷生成的活化能分布为Gaussian分布,采用指定的指前因子,结合碳同位素测定解释了热解甲烷的生成机理.
热重分析是研究复杂材料,如煤、干酪根及生物质等热解动力学的常用方法,其化学动力学模型有总包反应动力学模型、分段一级反应模型、最大反应速率模型、平行一级反应模型及分布活化能模型(DAEM)、Friedman反应模型;4基于结构的煤热解脱挥发份动力学模型主要有FLASHCHAIN模型、5-11官能团-脱聚、蒸发、交联模型(FG-DVC)12,13及化学渗透脱挥发份模型(CPD).14,15由于分布活化能模型认为煤中的化学键能符合某一种分布,煤的热解活化能也符合其分布,因此,能将热解活化能与煤中各种化学键能联系起来;同时,由于将至少三种升温速率下的失重曲线结合起来进行动力学处理,减小了升温速率对动力学参数求解的影响,较其它方法具有较多优点.Arenillas等16应用n(n=0, 1,2,3)级动力学方程、FG-DVC模型及分布活化能模型对煤的脱挥发份动力学进行对比分析,也证实了这一点.DAEM的首次提出是为了描述金属膜的电阻变化,17经过长期的发展,DAEM在复杂有机质热解反应动力学研究方面取得了很大进展,建立了一系列处理方法,如阶跃近似法、18拐点切线法、19Miura微分法18和Miura积分法20等,前两种方法针对单一失重曲线进行讨论,后两种方法将不同升温速率下的失重曲线联系起来处理,实现了数据处理方式上的重要进步.另外,Miura积分法由于不需要预先假定活化能分布函数和指定指前因子,因此,更加适合于复杂有机质的热解动力学分析,目前已广泛用于沉积有机质、生物质及垃圾废弃物热解动力学过程21-26及热解过程中挥发份的生成动力学.25,26
本文采用分布活化能模型对伊敏褐煤丝炭腐植酸及脱灰丝炭腐植酸的热解过程及氢气的析出过程进行分析,重点考察腐植酸在热力作用下的缩合反应机制及其动力学过程,探讨其化学反应的实质及其过程,以期加深对煤化作用化学机制及煤热解过程中氢气生成机制的认识.
2 实验及模型
2.1 煤样选取及样品制备方法
煤样采自内蒙古海拉尔盆地伊敏露天矿区,时代为早白垩世,丝炭样品采用手选剥离获得.样品在研钵内研磨粉碎后过筛,粒度≤74 μm,80°C下真空干燥2 h后保存备用.煤样脱灰处理参照文献,27腐植酸的提取参照文献,28所用的化学试剂HCl、HF和NaOH均为分析纯(AR).原煤丝炭(fusain,简称F)及脱灰丝炭(demineralized fusain,简称DF)的工业分析和元素分析按GB/T212-1991标准进行分析;伊敏褐煤丝炭腐殖酸(F-HA)和脱灰丝炭腐殖酸(DF-HA)的工业分析按GB/T212-1991标准进行,元素分析采用微量分析方法得到,氧元素含量均采用差减法获得,其结果见表1.同时样品的H/C和O/C原子比也列于表1.从表1可以看出,脱灰后煤中的灰分从6.92%下降为0.30%,因此脱灰效果良好.脱灰后碳含量增高,而其它元素则降低,其原因可能是在元素分析过程中,一些无机物质影响元素分析的结果.两种腐植酸样品氧含量很高,碳含量较低,氮含量较原提取煤样都有不同程度的升高,这也许与其含有较多的-COOH、-OH、醚键、苯醌及-NH等官能团有关.29
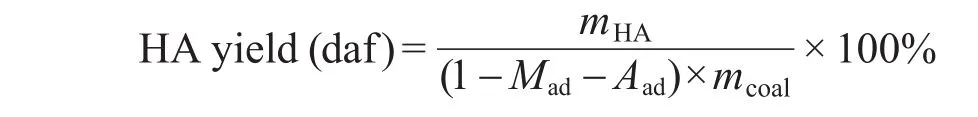
HA提取率采用下式计算:计算得到伊敏F-HA提取率为14.2%,DF-HA提取率为16.9%.丝炭脱灰后HA提取率比脱灰前增加2.7%,其原因可能是煤中的阳离子主要与煤结构中的-COOH、-OH、-SH及-NH等极性官能团发生相互作用,脱灰过程破坏了有机-无机间相互作用,29-31特别是高价阳离子,如Fe3+、Al3+等,与煤大分子结构的相互作用对HA提取率影响较大.
2.2 热重-质谱(TG-MS)实验
TG-MS采用法国Setaram公司的TGA92热重分析仪和瑞士Balzers公司的四极滤质质谱仪QMS422联用装置.干燥基下称重样品10 mg,从常温分别以5、20和50°C·min-1的升温速率升到1000°C,实验气氛为Ar,气体流速为45 mL·min-1.
为了减少系统波动带来的误差,对所得电信号的漂移进行了校正;同时,由于每种挥发份具有其独自的仪器响应因子,因此对挥发份的生成速率曲线进行了归一化处理.归一化方法参照文献,32经过处理后不同样品的挥发份生成速率曲线具有可比性.
2.3 分布活化能模型
分布活化能模型基于如下两点假设:(1)热解过程由许多相互独立的一级不可逆反应组成,即无限平行一级反应假设;(2)每个反应有确定的活化能(E)值,所有反应的E值呈某种连续分布,即活化能分布假设.

本文按照Miura积分法21进行计算.煤的热解过程满足下式:式中:V*为热解反应挥发份的总析出量,V为某一温度T时挥发份的总析出量,则V/V*为热解反应产物某一温度T时的转化率;k0为指前因子;f(E)为活化能分布函数;E为活化能;R为气体常数,R=8.314 J· mol-1·K-1;T为热解温度,单位为K.
其中,活化能分布函数f(E)满足

公式(1)可简化为
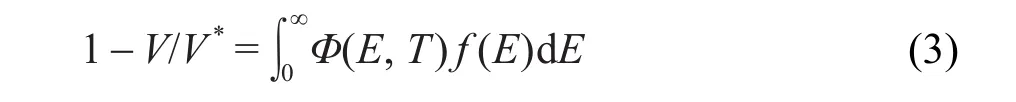
这里,
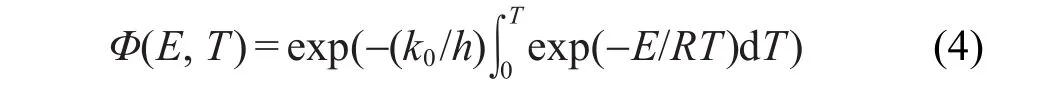
其中,T=T0+ht,h为升温速率,t为升温加热时间,h= dT/dt.
式(4)可简化为
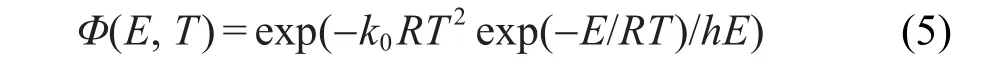
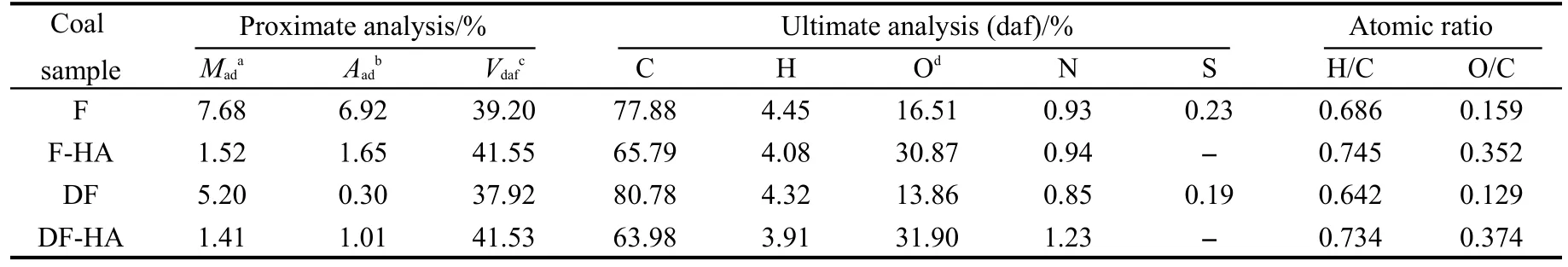
表1 煤样品的工业分析和元素分析Table 1 Proximate and ultimate analyses of coal samples
为了计算简便,可用一阶梯函数替代式(5),此函数有一特殊的活化能值Es

Es是经验值,当Φ(E,T)≅0.58时,存在
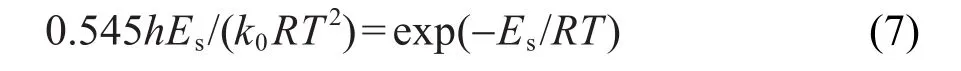
结合式(3)与(6),得
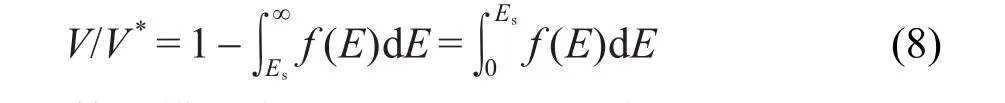
在该简化模型中,Arrhenius方程可描述为
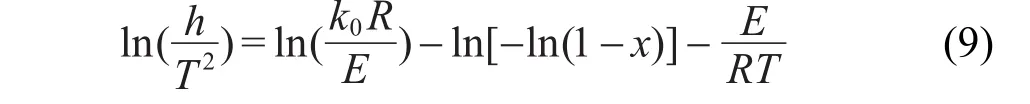
根据式(9)进行动力学分析的步骤主要包括:(1)实验测定不同升温速率下的失重曲线;(2)根据各失重曲线数据做ln(h/T2)-1/T曲线;(3)在ln(h/T2)-1/T图上作不同升温速率h条件下,同一转化率的Arrhenius直线,此直线斜率即为-E/R,由此可求得不同转化率下的E值;(4)本文不做任何活化能分布函数和指前因子的假定,活化能分布函数由式(8)通过d(V/ V*)/dEs得到,指前因子可通过式(9)得到.
3 结果与讨论
3.1 腐植酸的热解特征及动力学分析
3.1.1 热失重分析
图1(a)和1(b)分别为F-HA和DF-HA在不同升温速率下的热重-微分热重(TG-DTG)曲线,由曲线可以获得样品热解的特征参数,见表2.其中热解初始温度Ti和热解结束温度Te的计算方法参考文献33进行.从表2可以看出,在5、20和50°C·min-1三种升温速率条件下,F-HA的热失重Δw依次为41.4%、41.3%和41.2%,DF-HA依次为43.0%、42.3%和42.2%;两者相比,DF-HA相应的热失重稍大.随着升温速率的提高,两种腐植酸Δw均略有减少,F-HA差别较小;Ti和Te均向高温方向移动,热解温度区间减小;最大失重速率对应的温度TDTGmax延迟,最大失重速率(dw/dT)max减小.这是由于升温速率提高,样品经历的反应时间缩短,反应均需在较高的温度下才能进行,对热解产生了影响.相同升温速率下,DF-HA的Δw较F-HA的稍大;DF-HA的Ti和Te较F-HA的均稍有提前,Δ(Te-Ti)增大;(dw/ dT)max稍有减小,最大失重速率TDTGmax延迟.这可能意味着F-HA与原煤一样,存在着阳离子与含氧官能团的相互作用,这种作用将导致官能团的热解需要较高的能量,因此热解特征温度较高.
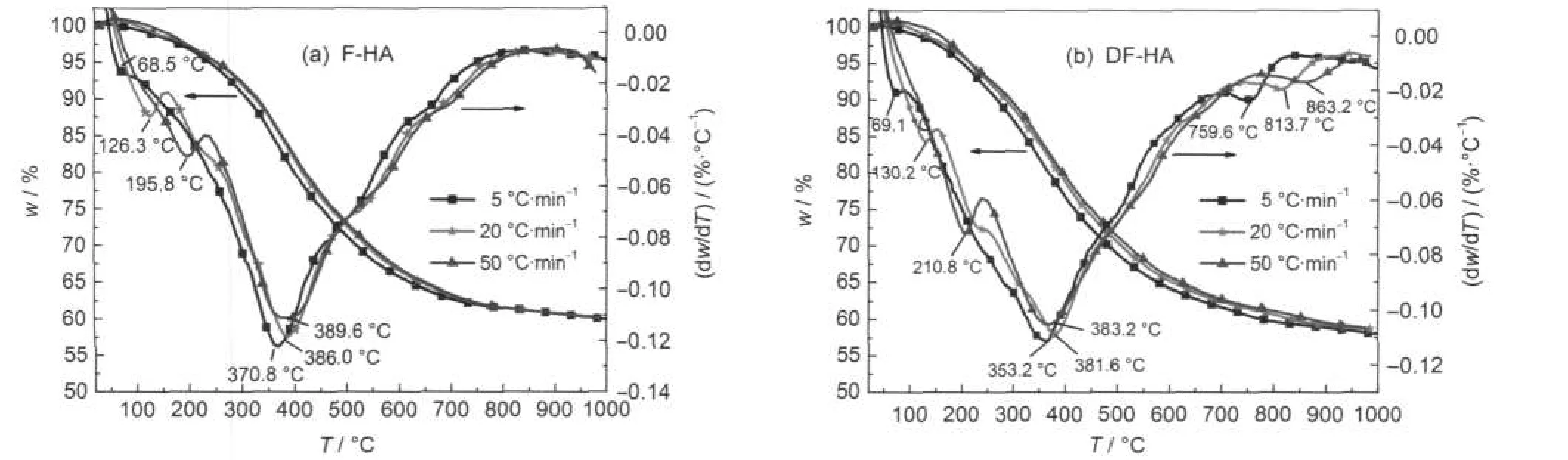
图1 F-HA(a)和DF-HA(b)在不同升温速率下的TG-DTG曲线Fig.1 Thermogravimetry-derivative thermogravimetry(TG-DTG)curves of F-HA(a)and DF-HA(b)at different heating rates
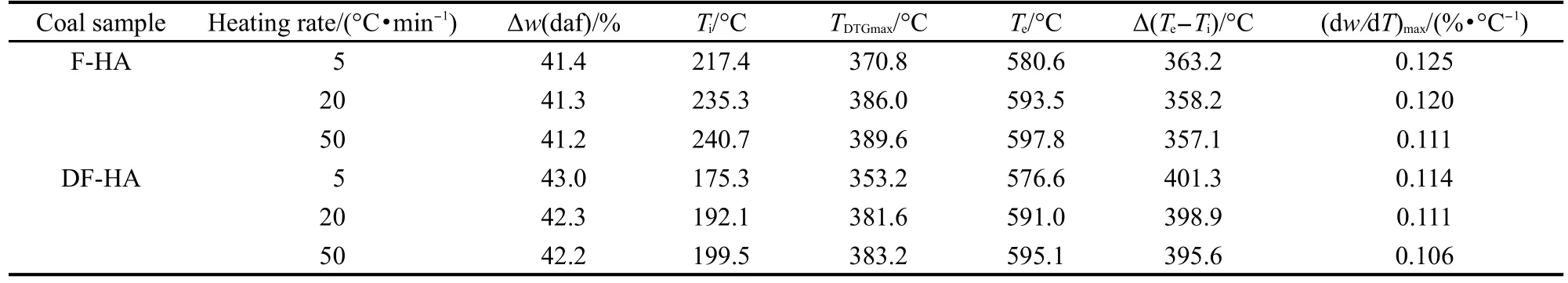
表2 腐植酸样品热解的特征参数Table 2 Pyrolysis characteristic parameters of HAsamples
由图1(a)和1(b)中可以看到,从DTG曲线上能够明显分辨出随着温度升高F-HA和DF-HA的热失重速率峰,为较好分辨化学反应类型温度范围提供一定依据.随着升温速率升高,F-HA依次在68.5、126.3和195.8°C处存在一个热解速率峰,DF-HA该峰相应出现在69.1、130.2和210.8°C,较F-HA相应温度点滞后.同时可以看到,随着升温速率的升高, F-HA依次在370.8、386.0和389.6°C达到最大热失重速率,而DF-HA最大热失重速率峰依次出现在353.2、381.6和383.2°C,较F-HA相应温度点提前.该阶段热失重较大,热失重速率也快.随着温度的进一步升高,从图1的DTG曲线上可以看到F-HA和DF-HA均存在两个平缓的热失重速率峰,其中, F-HA两个峰均较为明显,而DF-HA的第二个峰较小,该阶段为主热解阶段的延续.从图1(b)中DTG曲线可以看到,随着升温速率的升高,DF-HA依次在759.6、813.7和863.2°C存在一个明显的热失重速率峰,该峰在图1(a)F-HA的DTG曲线上并不存在.
3.1.2 腐植酸热解过程的活化能分布分析
3.1.2.1 热解活化能分布
图2为F-HA和DF-HA的ln(h/T2)与1/T在不同转化率下的关系曲线,是由热失重速率曲线通过选取转化率为0.01-0.96范围内一系列数据点做出ln(h/T2)与1/T的相关曲线得到,由此可以算出不同转化率下的活化能E和指前因子k0.图3为不同升温速率下转化率V/V*与温度T的关系曲线,可以看到腐植酸热解转化率随温度升高的变化情况.图4为F-HA和DF-HA的转化率V/V*与活化能E的关系曲线,可以看到转化率随着热解活化能的变化情况.根据式(8)通过d(V/V*)/dEs得到了活化能分布函数f(E),并将其与标准Gaussian分布进行了对比,结果见图5.
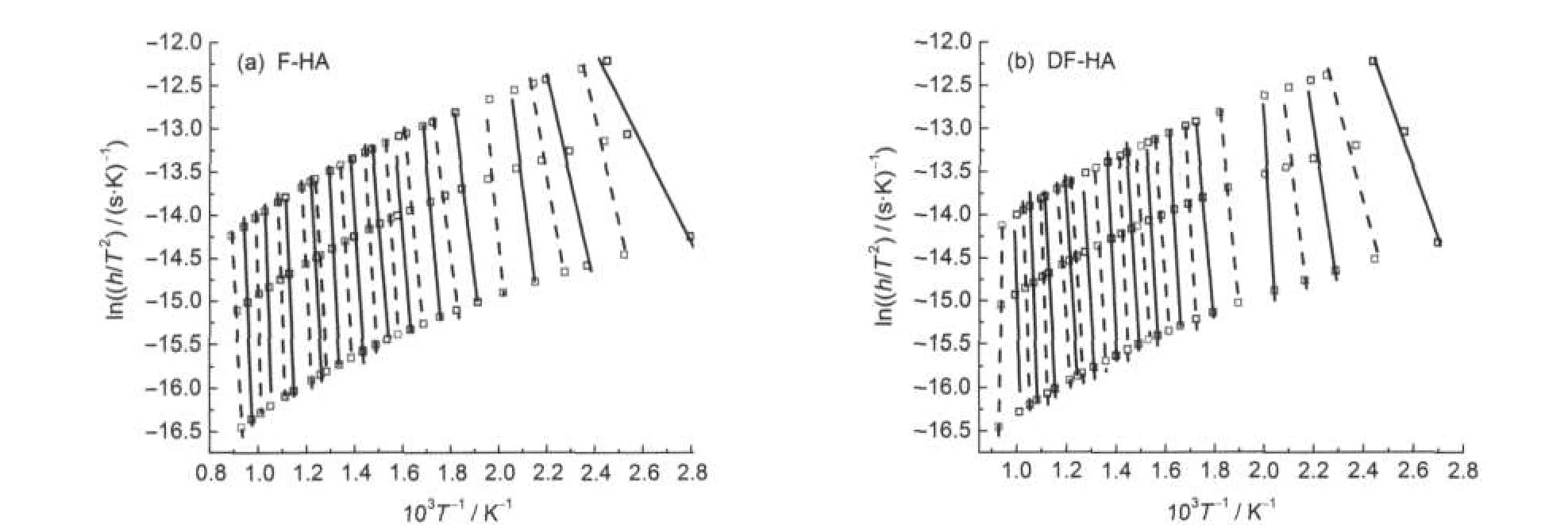
图2 ln(h/T2)与1/T在不同转化率(V/V*)下的关系曲线Fig.2 Relationship curves of ln(h/T2)vs 1/T at selected conversion ratio values(V/V*) (a)V/V*(from right to left):0.01,0.03,0.05,0.07,0.10,0.15,0.20,0.25,0.30,0.35,0.40,0.45,0.50,0.55,0.60,0.65,0.70,0.75,0.77,0.80, 0.85,0.87,0.90,0.92,0.94,0.96;(b)V/V*(from right to left):0.01,0.03,0.05,0.07,0.10,0.15,0.20,0.25,0.30,0.35,0.40,0.45,0.50,0.55, 0.60,0.65,0.70,0.75,0.77,0.80,0.85,0.87,0.90,0.92,0.94,0.96
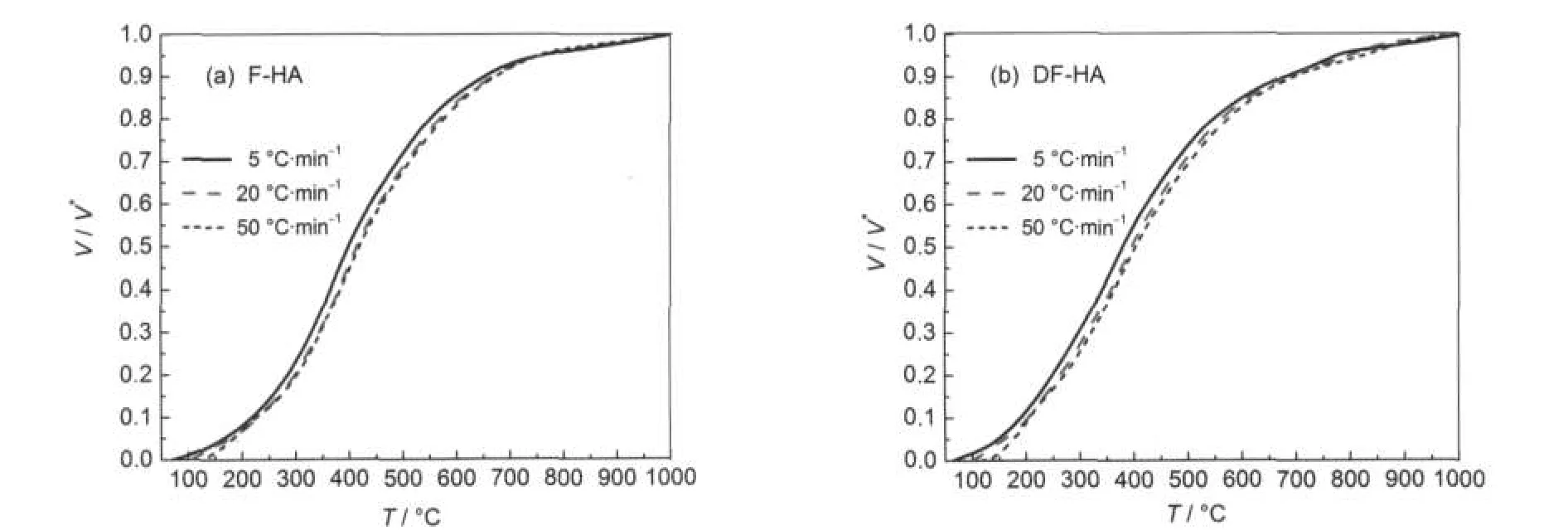
图3 不同升温速率下V/V*与T的关系曲线Fig.3 Relationship curves of V/V*vs T at different heating rates
从计算结果整体来看,F-HA热解活化能分布范围为66.9-816.0 kJ·mol-1,平均活化能E0为400.6 kJ·mol-1,标准偏差σ为180.7 kJ·mol-1,指前因子k0为;DF-HA热解活化能范围为45.8-784.3 kJ·mol-1,平均活化能E0为357.7 kJ·mol-1,标准偏差σ为185.1 kJ·mol-1,指前因子k0为1011-1039s-1.可见,两种腐植酸热解活化能分布范围均较宽,且σ值越大,表明活化能的数值越分散;与F-HA相比,DF-HA热解平均活化能E0较小,而σ值较大.从图4也可以看到,F-HA与DF-HA相比,当转化率V/ V*<0.75时,DF-HA活化能值较小;当转化率V/V*>0.75时,两者活化能值差别不大,F-HA活化能值稍小.孙佰仲等[24]也发现矿物质催化作用改变了油页岩脱挥发分过程中络合物的性质,并使活化能有所降低.煤结构研究表明,34,35在低煤级阶段存在阳离子-含氧官能团相互作用,而随着煤级的增高,这种作用将减小,但存在阳离子-π作用.而伊敏丝炭为褐煤,煤阶较低,主要存在阳离子-含氧官能团相互作用,通过酸洗脱灰处理,能在很大程度上减小这种作用.30,31脱灰前后丝炭提取出的腐植酸不但结构存在差异,而且也会存在这种相互作用,脱灰处理将导致这种作用明显减弱,从而引起脱灰煤HA热解平均活化能较小.
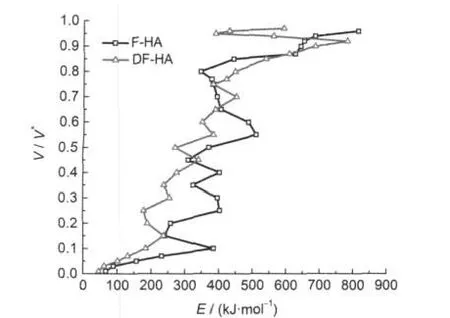
图4 转化率V/V*与活化能E的关系曲线Fig.4 Relationship curves of V/V*vs E
3.1.2.2 热解过程的阶段划分及其化学反应
从图4可以看到,热解过程活化能的整体趋势是随着转化率的升高而增加,但也表现出阶段性集中分布的特征.从图5可以看到,F-HA热解活化能分布函数f(E)具有类Gaussian分布形式和阶段性集中分布特点,且具有一定的对称性,主峰位与标准Gaussian分布相一致;DF-HA活化能分布函数f(E)呈类Gaussian分布形式,主峰位与Gaussian分布相比,偏向低活化能值.这应该与两种腐植酸中化学键的键能分布情况相关.根据图3、图4和图5,结合腐植酸的热失重特征,可将F-HA热解过程划分为四个阶段,DF-HA热解过程划分为五个阶段,各阶段的活化能值分布情况见表3.
图6为腐植酸样品在升温速率为20°C·min-1条件下热解过程中低级烃类C1-C4的逸出曲线,图7为腐植酸样品在升温速率为20°C·min-1条件下热解过程中H2、氧化物及小分子芳烃的逸出曲线,结合腐植酸热解过程的动力学计算及热失重特征,对腐植酸热解过程各阶段化学反应进行分析.
第I阶段:为干燥脱水、脱气及脱除不稳定羧基阶段.F-HA和DF-HA活化能分布主峰分别为66.9和142.4 kJ·mol-1,主要以分子间及分子内氢键的破坏及不稳定含氧官能团,如甲氧基、不稳定羧基等的热解为主,从而导致H2O在255.0°C附近存在较大的逸出宽峰,CH4在该阶段存在一逸出峰,CO2大量逸出.已有的研究4表明,甲氧基、不稳定羧基是地质有机质中较早热解的官能团,在温度较低时就能热解断裂,生成甲烷、水和二氧化碳.图1中DTG曲线低温下存在的一个明显热解速率峰,为这些反应共同作用的结果.
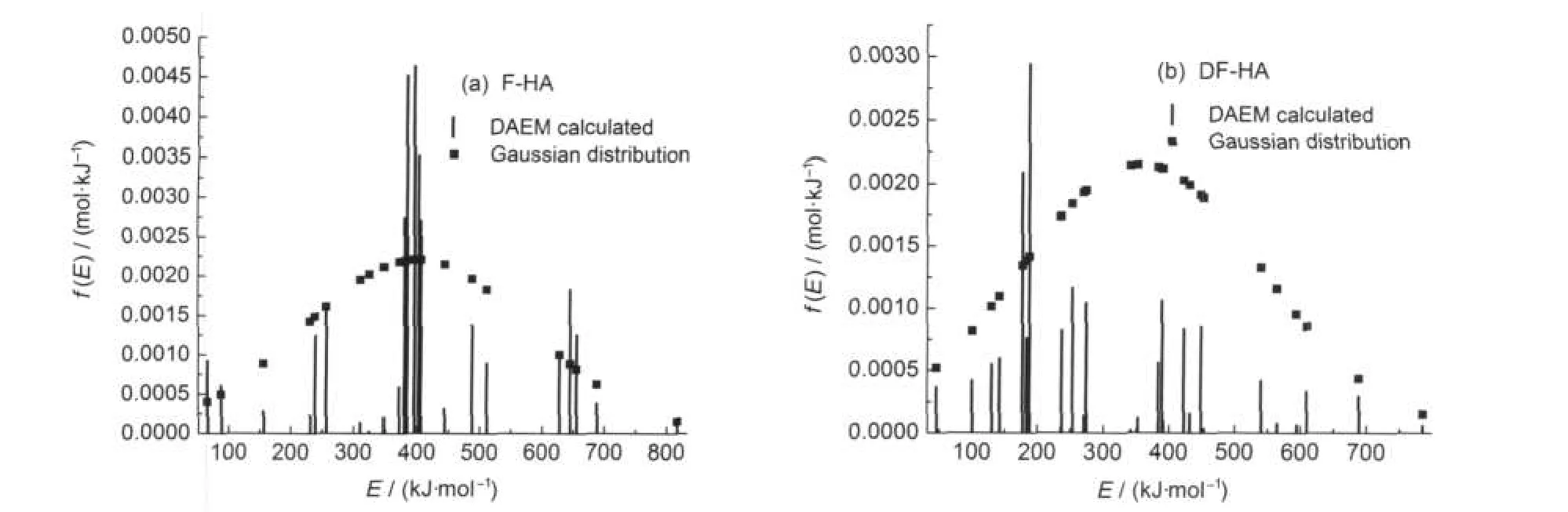
图5 HA热解过程的活化能分布函数f(E)与标准Gaussian分布Fig.5 Distribution functions of activation energy f(E)and the standard Gaussian function of HAduring pyrolysis
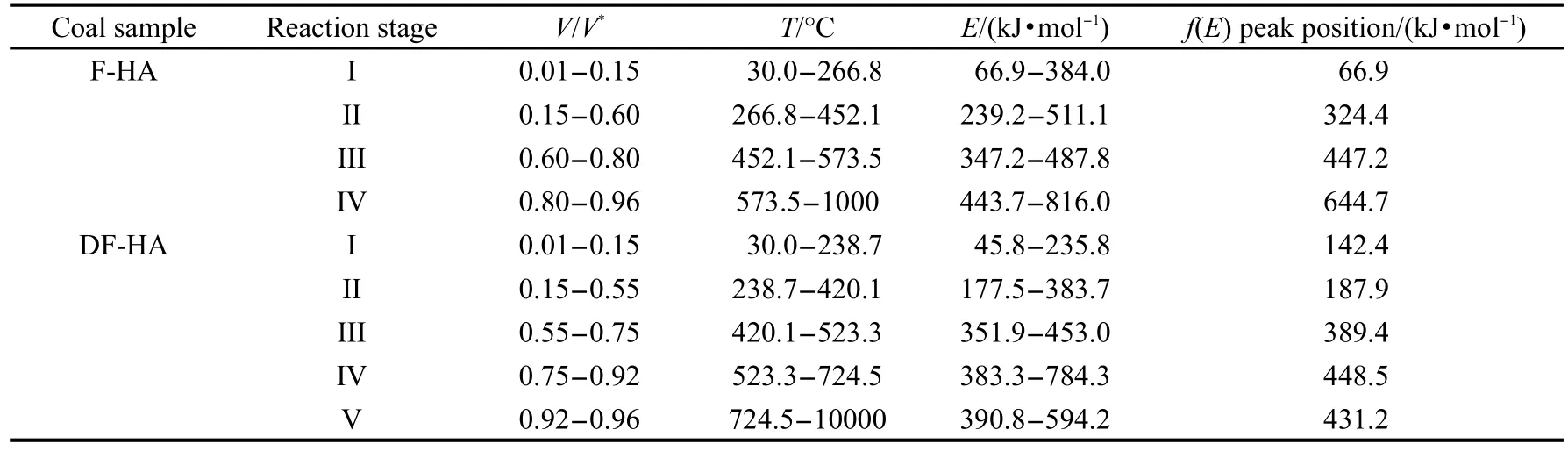
表3 腐植酸样品热解过程不同阶段的动力学参数值Table 3 Kinetic values of different stages for HAsamples during pyrolysis
图6 腐植酸样品在升温速率为20°C·min-1条件下热解过程中C1-C4低级烃类的逸出曲线Fig.6 Evolution curves of low rank hydrocarbon C1-C4at a heating rate of 20°C·min-1during pyrolysis of HAsamples
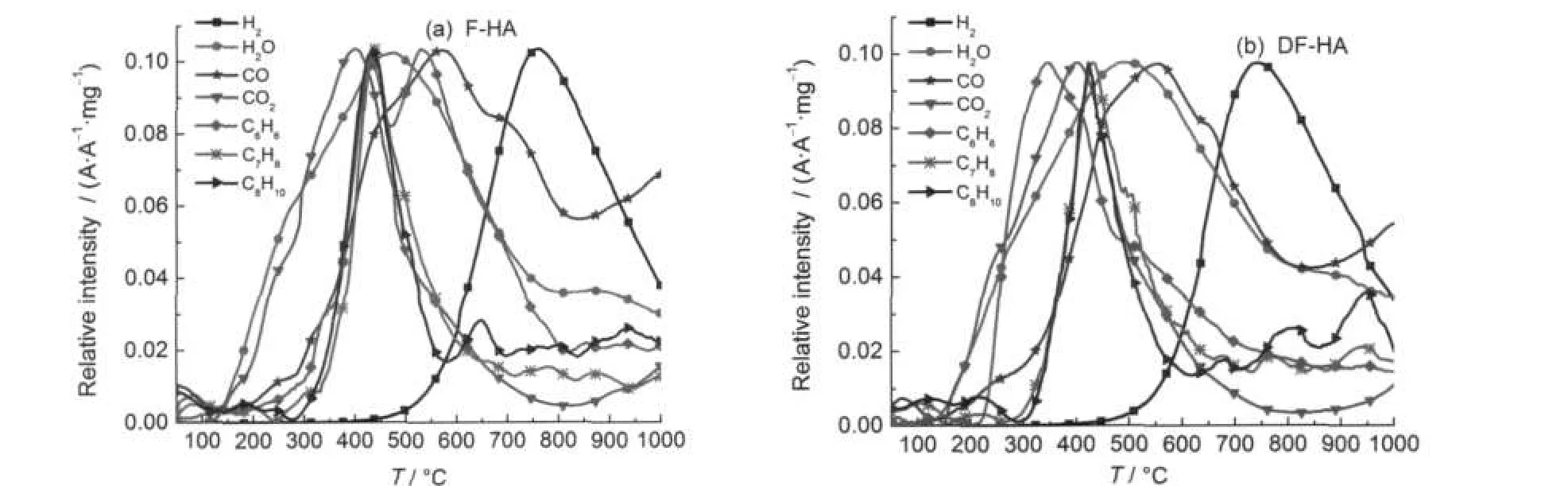
图7 腐植酸样品在升温速率为20°C·min-1条件下热解过程中H2O、CO、CO2、H2及小分子芳烃的逸出曲线Fig.7 Evolution curves of H2O,CO,CO2,H2,and small molecular aromatic hydrocarbons at a heating rate of 20°C·min-1 during pyrolysis of HAsamples
第II阶段:该阶段热失重量较大,热失重速率达到最大.F-HA和DF-HA温度范围依次为266.8-452.1°C和238.7-420.1°C,为腐植酸主热解的活泼热分解初期阶段,主要发生解聚和分解反应.F-HA和DF-HA热解过程的产物,以C3H6、+C4H7和+C4H9为代表的长链烃类峰温分别为421.8和 412.5°C,峰形均为尖锐的单峰;而C2H6和+C2H2短链烃类峰温分别为445.2和439.5°C;低级小分子芳烃,C6H6逸出为双峰,第一个逸出峰的峰温分别为432.6和344.3°C,C7H8和C8H10的峰温分别为436.9和428.0°C.由此可见,C2-C4与低级小分子芳烃的逸出峰温均在412.5-445.2°C之间,说明均来自煤大分子结构降解和裂解统一过程,且主要与连接芳香结构体系的脂肪类交联键的热解断裂有关.这是由于与芳香结构相连的α位断裂比较困难,活化能较高,而脂肪类交联键的热解断裂则较容易;36芳香烃类物质的生成主要与连接芳香结构体系的脂肪类交联键的热解断裂有关,同时生成小分子脂肪烃类.再者,腐植酸含有大量羧基、羟基和醚键官能团,该阶段也为脱羧、脱羟基和醚键发生断裂的主要阶段,伴随有大量CO2、H2O和CO生成.37从图7可以看到F-HA和DF-HA中主要含氧化合物在该阶段的逸出情况,CO2分别在398.1和399.0°C存在很大的逸出峰,为热解CO2主要析出阶段.CO存在一逸出肩峰,H2O的逸出强度也很大.另外,在这阶段也可能存在热解形成的烷基自由基的二次热解.从图4也可看出,F-HA和DF-HA活化能分布范围分别为239.2-511.1 kJ·mol-1和177.5-383.7 kJ·mol-1,主峰分别为324.4和187.9 kJ·mol-1,这是由于该阶段存在复杂的化学反应,因此活化能值波动均较大.
第III阶段:该阶段活化能值较为集中,随着转化率的升高,活化能持续减小,该阶段为主热解阶段的延续,从图1中DTG曲线可以看到在该阶段存在一宽缓的失重速率峰.F-HA和DF-HA活化能值范围差别不大,但其主峰值相差较大,分别为447.2和389.4 kJ·mol-1.CO分别在567.6和548.8°C出现最大逸出峰,主要由羰基高温下的断裂生成,同时CO还可与煤中的氧杂原子结合,生成CO2.38H2O分别在471.2和489.1°C出现最大逸出峰,为HA中含氧官能团(主要为酚羟基)生成的热解水.CH4峰温分别为554.5和562.1°C,C6H6分别在527.8和504.8°C出现第二个逸出峰,较甲烷稍早,H2在该阶段开始逸出,由此可推测该阶段苯的生成为在氢自由基存在下与苯类物质相连的甲基直接脱落生成C6H6,同时生成CH4;也不排除环烷烃芳构化作用的贡献,生成苯的同时生成H2.
第IV阶段:该阶段活化能值最大,F-HA活化能高达816.0 kJ·mol-1,主峰活化能为644.7 kJ·mol-1; DF-HA为784.3 kJ·mol-1,主峰为448.5 kJ·mol-1.该阶段以缩聚反应为主,热解活化能值较大,主要生成H2和CO,其次为H2O及CH4和少量CO2.CO在700.0°C附近出现一逸出峰,可能与较稳定的含氧杂环、苯醌及酮等断裂有关.苯的生成为其第二个逸出峰的延续,可能为联苯类物质连接键断裂生成,所需活化能值较大.该阶段分别与图1(a)和1(b) DTG曲线在590.3-830.1°C和530.5-710.2°C存在的失重速率峰相对应.
对于DF-HA而言,从图4可以看到,存在第V阶段,转化率V/V*>0.92,DF-HA活化能值先减小后又增加,活化能范围为390.8-594.2 kJ·mol-1,主峰为431.2 kJ·mol-1.这与图1(b)中DTG曲线在高温下存在的一个明显失重速率峰相对应,可能和原先与金属离子呈螯合状态存在的多环烃类结构单元脱灰后被释放出来高温下发生热缩聚作用有关.
3.2 热解氢气生成特征及动力学分析
3.2.1 热解氢气生成特征
图8为热解氢气在不同升温速率下的逸出曲线,为了便于对比分析,曲线经过了归一化处理,其不同升温速率下的热解生成特征参数见表4.同一升温速率下,DF-HA热解氢气开始析出温度T0稍有提前,而析出峰温Tmax提前较多.热解过程中氢气的总生成量5°C·min-1时较少,其余升温速率下差别不是太大.随着升温速率的提高,开始析出温度T0和析出峰温Tmax均滞后.H2从接近400°C开始大量析出,直至热解终温仍有析出,热解生成温度段较长.
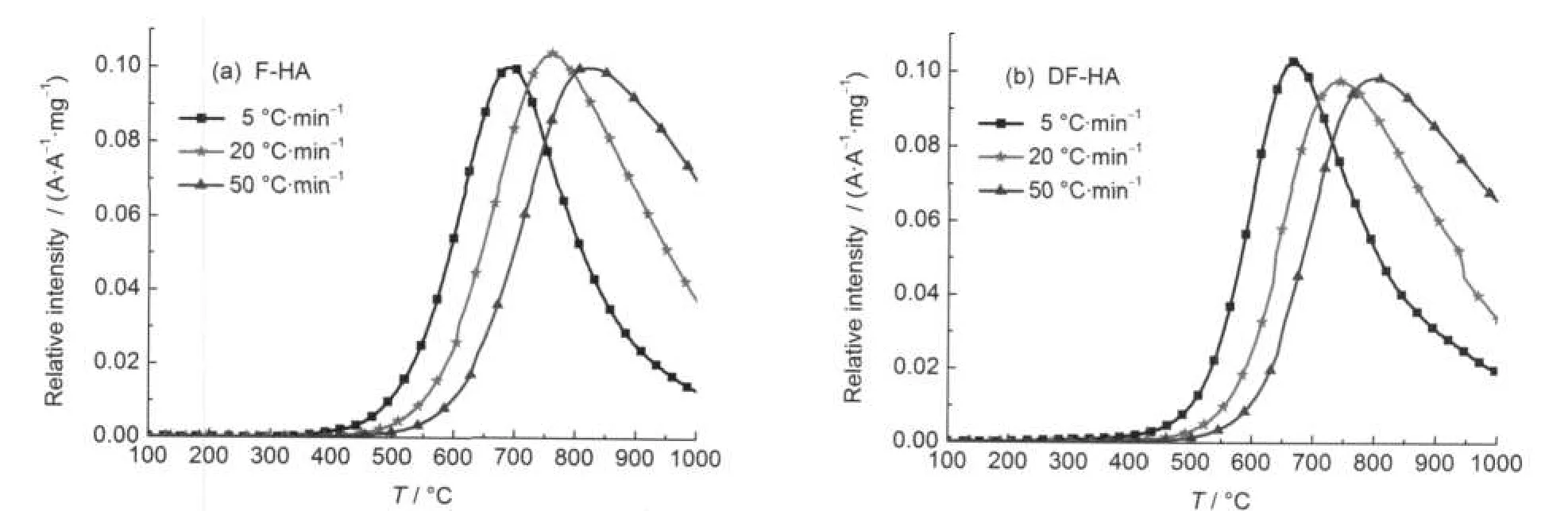
图8 热解氢气在不同升温速率下的逸出曲线Fig.8 Evolution curves of hydrogen at different heating rates during pyrolysis
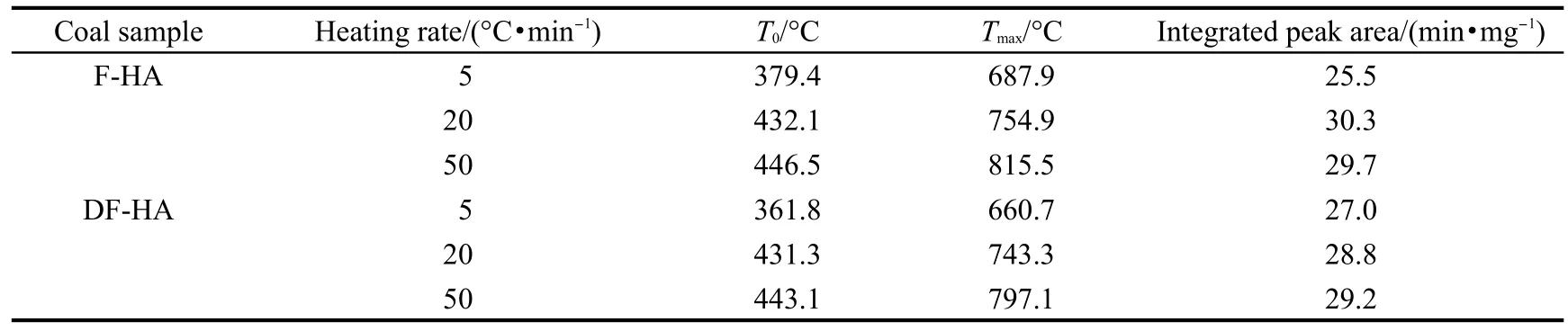
表4 热解氢气生成的特征参数Table 4 Pyrolysis characteristic parameters of hydrogen generation
3.2.2 热解氢气生成的活化能分布及氢气的生成阶段
3.2.2.1 热解氢气生成动力学参数
图9为热解氢气在不同V/V*下的Arrhenius曲线,是通过选取F-HA和DF-HA热解氢气转化率分别为0.01-0.92和0.01-0.90范围内一系列数据点做出ln(h/T2)与1/T的相关曲线得到,由此可以算出热解氢气生成在不同转化率下的活化能E和指前因子k0.图10为不同升温速率氢气生成转化率V/V*与温度T的关系曲线,可以看到氢气生成转化率随温度升高的变化情况.图11为氢气生成转化率V/V*与活化能E的相关关系,可以看到随着转化率的升高,活化能整体呈增大趋势.
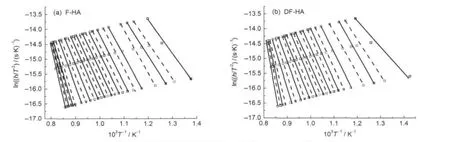
图9 热解氢气在不同转化率V/V*下的Arrhenius曲线Fig.9 Arrhenius plots of hydrogen evolution at selected V/V*values (a)V/V*(from right to left):0.01,0.02,0.03,0.05,0.10,0.15,0.20,0.25,0.30,0.35,0.40,0.45,0.50,0.55,0.60,0.65,0.70,0.75,0.80,0.85, 0.87,0.88,0.90,0.91,0.92;(b)V/V*(from right to left):0.01,0.02,0.03,0.05,0.10,0.15,0.20,0.25,0.30,0.35,0.40,0.45,0.50,0.55,0.60, 0.65,0.70,0.75,0.80,0.85,0.87,0.88,0.90
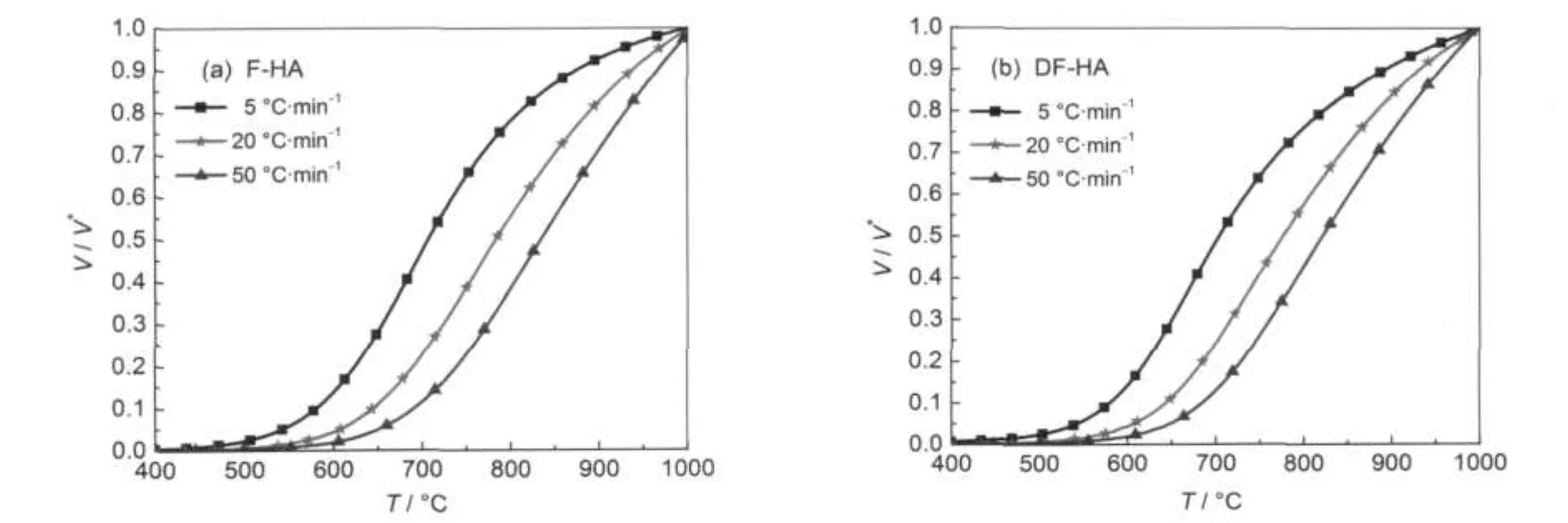
图10 不同升温速率下氢气生成转化率V/V*与温度T的关系曲线Fig.10 Relationship curves of V/V*vs T for hydrogen generation at different heating rates
图12为活化能分布函数f(E)与标准Gaussian分布曲线,其中活化能分布函数f(E)由活化能E与转化率V/V*的关系曲线根据式(8)通过d(V/V*)/dEs得到,从图中可以发现,F-HA与DF-HA热解氢气生成活化能的分布形式类似,均为类高斯分布,其主峰位与标准高斯分布相差较大,且两种腐殖酸的主峰位大致相同.F-HA热解氢气生成活化能范围为92.5-316.0 kJ·mol-1,E0为166.1 kJ·mol-1,指前因子k0为,σ为56.9 kJ·mol-1;DF-HA热解氢气生成活化能范围为67.1-371.4 kJ·mol-1,E0为180.9 kJ·mol-1,指前因子k0为104-1014s-1,σ为80.4 kJ· mol-1.两者相比DF-HA的E0和σ值均较大,平均活化能E0值较大,说明DF-HA热解生成氢气整体所需活化能较大;σ值偏大,说明DF-HA热解氢气生成相关反应的活化能分布较为分散.
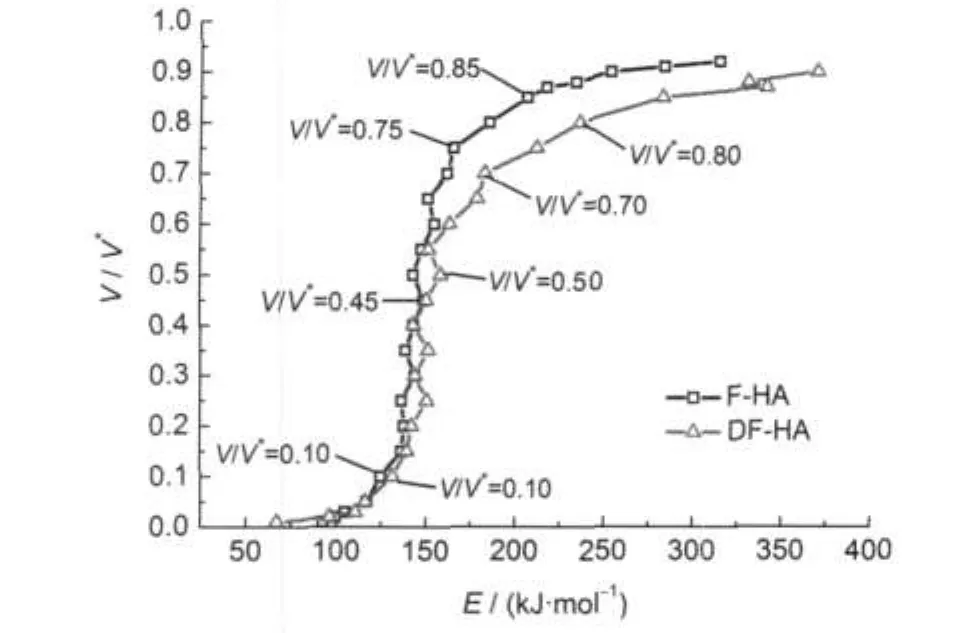
图11 氢气生成转化率V/V*与活化能E的相关关系Fig.11 Relationships of V/V*vs E for hydrogen generation
图13为lnk0和E的相关关系,可以看出lnk0与E存在良好的线性相关性,相关系数均大于0.99.k0与E存在补偿效应,所以在腐植酸热解过程中k0不能设定为常数.k0与E满足:k0=αeEβ(α,β为常数),F-HA热解氢气α=e-3.5325,β=0.0983;DF-HA热解氢气α= e-3.8274,β=0.1016.
3.2.2.2 热解氢气生成阶段及机制分析
从图11可以看到当V/V*=0.55时,为两种腐植酸热解氢气生成活化能相对大小的转折点.从图10可知,此时热解氢气的生成温度,F-HA为799.5°C, DF-HA为793.1°C.当V/V*<0.55时,F-HA热解氢气生成活化能值较DF-HA稍小,两者差别不大;当V/ V*>0.55时,F-HA活化能值较DF-HA大大减小.说明矿物质的存在对缩聚脱氢反应具有促进作用,矿物质脱除后,煤的有机物理结构发生了变化,对提取出的腐植酸热解氢气生成产生了影响,这与两种腐植酸整体热解过程中以发生缩聚反应生成H2为主的第IV阶段脱灰前后HA热解活化能值相对高低相一致.再者,Faure等39的研究表明,Na-蒙脱石的存在有利于芳香化合物的生成.因此,F-HA热解氢气生成活化能较DF-HA低的原因就在于无机金属离子的存在有利于芳构化作用.
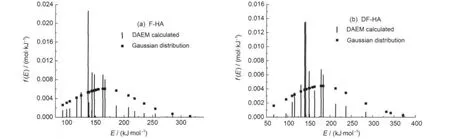
图12 氢气生成活化能分布函数f(E)与标准Gaussian分布Fig.12 Distribution functions of activation energy f(E)and the standard Gaussian distribution for hydrogen generation
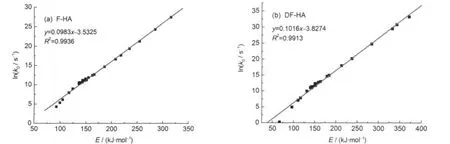
图13 lnk0和E的相关关系Fig.13 Relationships of lnk0vs E
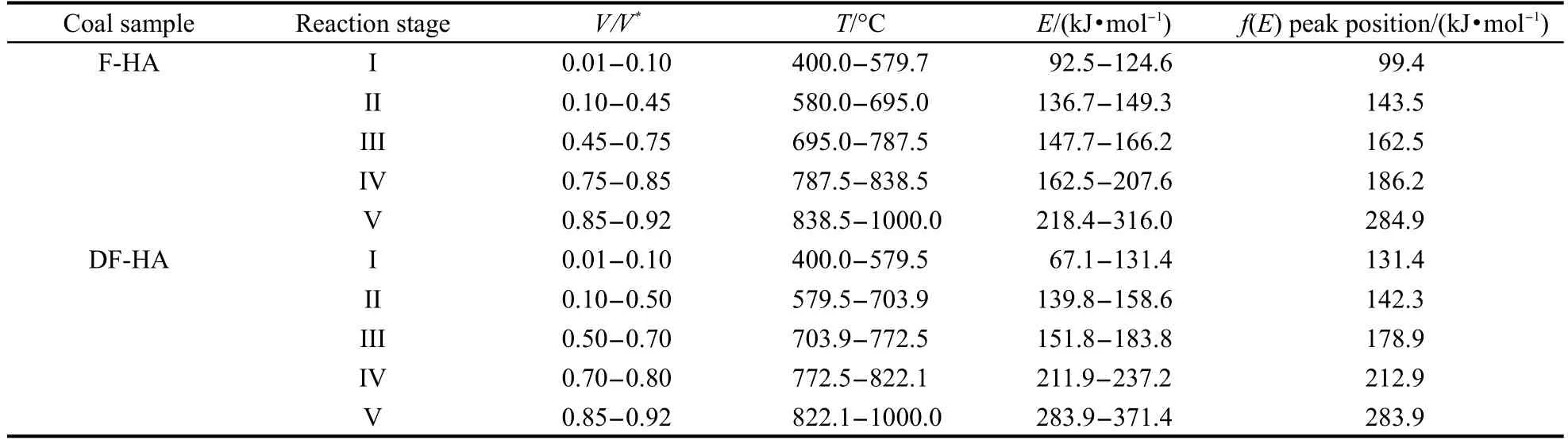
表5 腐植酸样品热解氢气生成不同阶段的动力学参数值Table 5 Kinetic values of different hydrogen evolution stages for HAsamples
结合图10、图11和图12可以将F-HA和DF-HA热解氢气的生成均划分为五个阶段,各阶段的划分情况见表5.
活化能分布的阶段性集中现象反映了氢气生成的不同化学反应机制.热解氢气生成的早期阶段主要为热解生成的较长链脂肪烃类二次热解产生的氢自由基间的结合、链烷烃的环化、环烷烃的芳构化、氢化芳香结构脱氢,2,40,41结合反应温度及活化能值,该类反应主要发生在热解氢气生成的前两个阶段.此时,F-HA和DF-HA反应温度均低于695.0和703.9°C,生成活化能分别小于149.3和158.6 kJ· mol-1.第III阶段F-HA和DF-HA温度范围分别为703.9-772.5°C和695.0-787.5°C,从图8可以看到,氢气的最大生成速率峰温位于该温度段内;两种HA活化能值主峰分别为162.5和178.9 kJ·mol-1,为HA芳香结构发生热缩聚反应的早期阶段.氢气生成最后两个阶段,生成温度较高,活化能值较大, F-HA两阶段主峰依次为186.2和284.9 kJ·mol-1, DF-HA两阶段主峰依次为212.9和283.9 kJ·mol-1,主要为芳香体系继续稠合、增大和芳香层片叠加过程脱氢.41
4 结论
(1)伊敏煤丝炭原煤的HA提取率为14.2%,而脱灰丝炭为16.9%,脱灰后HA提取率增加.
(2)F-HA与DF-HA相比,DF-HA的热解活化能E0较小,σ值较大;转化率V/V*<0.75时,F-HA活化能值较小;当V/V*>0.75时,两者活化能值差别不大.这表明酸洗脱灰减弱了HA中阳离子-含氧官能团间相互作用.
(3)热解氢气生成的动力学表明,DF-HA的E0和σ值均较大.热解氢气生成转化率V/V*<0.55时, F-HA氢气生成活化能值较DF-HA稍小;当V/V*>0.55时,F-HA活化能值较DF-HA大大减小.F-HA热解氢气生成活化能较DF-HA低的原因在于无机金属离子的存在有利于芳构化作用.
(4)根据F-HA和DF-HA热解活化能分布函数f(E)的特征、转化率V/V*与温度T及活化能E的关系,结合腐植酸的热失重特征,将F-HA热解过程划分为四个阶段,DF-HA热解过程划分为五个阶段,并对腐植酸热解过程各阶段化学反应进行了分析.热解氢气生成过程可划分为五个阶段,反映了其生成的不同化学反应机制.
(1)Yang,Q.;Han,D.X.Coal Geology of China;Coal Industry Press:Beijing,1979;Vol.1,pp 14-15.[杨 起,韩德馨.中国煤田地质学.北京:煤炭工业出版社,1979:Vol.1,14-15.]
(2)Huang,D.F.;Qin,K.Z.;Wang,T.G.;Zhao,X.G.;Xu,Y.Q. Oil from Coal:Formation and Mechanism;Petroleum Industry Press:Beijing,1955;pp 5-29. [黄第藩,秦匡宗,王铁冠,赵锡嘏,许云秋.煤成油的形成和成烃机理.北京:石油工业出版社,1995:5-29.]
(3) Cramer,B.Org.Geochem.2004,35,379.
(4) Fu,J.M.;Qin,K.Z.Geochemistry of Kerogen;Science& Technology Press of Guangdong:Guangzhou,1995;pp 268-269,546-552.[傅家谟,秦匡宗.干酪根地球化学.广州:广东科技出版社,1995:268-269,546-552.]
(5) Niksa,S.;Kerstein,A.R.Energy Fuels 1991,5,647.
(6) Niksa,S.;Kerstein,A.R.Energy Fuels 1991,5,665.
(7) Niksa,S.;Kerstein,A.R.Energy Fuels 1991,5,673.
(8) Niksa,S.Energy Fuels 1994,8,659.
(9) Niksa,S.Energy Fuels 1994,8,671.
(10) Niksa,S.Energy Fuels 1995,9,467.
(11) Niksa,S.Energy Fuels 1996,10,173.
(12)Solomon,P.R.;Hamblen,D.G.;Carangelo,R.M.Energy Fuels 1988,2,405.
(13)Solomon,P.R.;Hamblen,D.G.;Carangelo,R.M.Combust. Flame 1988,71,137.
(14) Grant,D.M.;Pugmire,R.J.Energy Fuels 1989,3,175.
(15) Fletcher,T.H.;Kerstein,A.R.Energy Fuels 1990,4,54.
(16) Arenillas,A.;Rubiera,F.;Pevida,C.;Pis,J.J.J.Anal.Appl. Pyrolysis.2001,58,685.
(17) Vand,V.Proc.Phys.Soc.(London)1942,A55,222.
(18) Miura,K.Energy Fuels 1995,9,302.
(19)Hashimoto,K.;Miura,K.;Watanabe,T.AICHE J.1982,28, 737.
(20) Miura,K.;Maki,T.Energy Fuels 1998,12,864.
(21) Sonobe,T.;Worasuwannarak,H.N.Fuel 2008,87,414.
(22)Quan,C.;Li,A.M.;Gao,N.B.Waste Manage.2009,2353.
(23) Liu,X.G.;Li,B.Q.J.Fuel Chem.Technol.2001,29,54. [刘旭光,李保庆.燃料化学学报,2001,29,54.]
(24)Sun,B.Z.;Wang,H.G.;Guan,X.H.Chem.Eng.(China)2007, 35,26.[孙佰仲,王海刚,关晓辉.化学工程,2007,35,26.]
(25)Yan,J.H.;Zhu,H.M.;Jiang,X.G.;Chi,Y.;Cen,K.F. J.Hazard.Mater.2009,162,646.
(26) Heidenreich,C.A.;Yan,H.M.;Zhang,D.K.Fuel 1999,78, 557.
(27)Chen,W.M.Coal Sci.Technol.1994,22,40.[陈文敏.煤炭科学技术,1994,22,40.]
(28)Yang,J.H.;Chen,W.M.;Duan,Y.L.Chemical Examination Manual of Coal;Coal Industry Press:Beijing,2004;pp 584-592.[杨金和,陈文敏,段云龙.煤炭化验手册.北京:煤炭工业出版社,2004:584-592.]
(29) Swaine,D.Org.Geochem.1992,78,259.
(30) Sugano,M.;Mashimo,K.;Wainai,T.Fuel 1999,78,945.
(31)Steel,K.M.;Besida,J.;OʹDonnell,T.A.;Wood,D.G.Fuel Process.Technol.2001,70,171.
(32) Arenillas,A.;Rubiera,F.;Pis,J.J.J.Anal.Appl.Pyrolysis. 1999,50,31.
(33) Shen,X.The Kinetics Study of Different Thermal,Thermo Gravimetry Analysis and Nonisothermal Solid Phase; Metallurgical Industry Press:Beijing,1995;pp 68-70. [沈 兴.差热、热重分析与非等温固相反应动力学.北京:冶金工业出版社,1995:68-70.]
(34) Painter,P.C.;Opaprakasit,P.;Scaroni,A.Energy Fuels 2000, 14,1115.
(35) Opaprakasit,P.;Scaroni,A.W.;Painter,P.C.Energy Fuels 2002,16,543.
(36) Zeng,F.G.;Jia,J.B.Acta Phys.-Chim.Sin.2009,25,1117. [曾凡桂,贾建波.物理化学学报,2009,25,1117.]
(37) Zhu,X.D.;Zhu,Z.B.;Han,C.J.;Tang,L.H.J.East China Univ.Sci.Technol.2000,26,14. [朱学栋,朱子彬,韩崇家,唐黎华.华东理工大学学报,2000,26,14.]
(38) Sun,Q.L.;Li,W.;Chen,H.K.;Li,B.Q.J.China Univ.Min. Eng.2003,32,665.[孙庆雷,李 文,陈皓侃,李宝庆.中国矿业大学学报,2003,32,665.]
(39) Faure,P.;Jeanneau,L.;Lannuzel,F.Org.Geochem.2006,37, 1900.
(40) Chen,C.X.;Sun,X.X.;Ma,L.Y.Prog.Nat.Sci.1995,5,83. [陈彩霞,孙学信,马毓义.自然科学进展,1995,5,83.]
(41) Porada,S.Fuel 2004,83,1191.
September 6,2011;Revised:October 31,2011;Published on Web:November 2,2011.
Kinetic Analysis of a Pyrolysis Process and Hydrogen Generation of Humic Acids of Yimin Lignite Fusain Using the Distributed Activation Energy Model
WANG Chuan-Ge ZENG Fan-Gui*PENG Zhi-Long LI Xia ZHANG Li
(Key Laboratory of Coal Science&Technology,Ministry of Education&Shanxi Province,Taiyuan University of Technology, Taiyuan 030024,P.R.China; Department of Earth Science&Engineering,Taiyuan University of Technology, Taiyuan 030024,P.R.China)
The thermal behavior of humic acids of fusain(F-HA)and humic acids of demineralized fusain(DF-HA)were investigated at heating rates of 5,20,and 50°C·min-1using an open system thermogravimetric analyzer coupled to a quadrupole thermogravimetry mass spectrometer(TG-MS).The pyrolytic and hydrogen generation kinetics were analyzed using the distributed activation energy model (DAEM)and the distribution functions of the activation energy were obtained.The results indicated:(1)the distribution function of the activation energy for F-HA has an asymptotic approximation to a Gaussian distribution during pyrolysis.Moreover,it has some symmetry properties and the same peak value as a standard Gaussian distribution.The distribution function of the activation energy for DF-HA has an asymptotic approximation to a Gaussian distribution during pyrolysis and its peak value is less than that of a standard Gaussian distribution.According to the conversion ratio vs temperature,activation energy relationships and the thermal mass loss features of humic acids,four and five stages of a general pyrolytic process were found for F-HA and DF-HA,respectively.The chemical reactions for each stage of the pyrolytic process are discussed in detail.(2)The distribution functions of the activation energy of hydrogen generation for F-HA and DF-HA have an asymptotic approximation to a Gaussian distribution during pyrolysis.The activation energy of hydrogen generation increased with an increase in the conversion yields but it also showed staged features.Based on the kinetic characteristics of their generation during pyrolysis their hydrogen generation processes can be divided into five stages,which reflect the different chemical reaction mechanisms.(3)The demineralization of Yimin fusain influences the thermal behavior, kinetics of the pyrolytic processes and the hydrogen generation of humic acid.
Humic Acid;Pyrolysis;Hydrogen generation;Distribution activation energy model; Kinetics; Fusain
10.3866/PKU.WHXB20122825
*Corresponding author.Email:zengfangui@tyut.edu.cn;Tel:+86-351-6010468.
The project was supported by the National Natural Science Foundation of China(41072116,40772097,40572094)and Specialized Research Fund for the Doctoral Program of Higher Education,China(20091402110002).
国家自然科学基金(41072116,40772097,40572094)和高等学校博士学科点专项科研基金(20091402110002)资助项目
O643;O641
