聚乙烯吡咯烷酮(PVP)和脲醛树脂(UF)包覆耐晒黄G的研究
2012-11-22李朝辉湛雪辉荀育军
李朝辉 湛雪辉,荀育军
(长沙理工大学化学与生物工程学院,中国 长沙 410076)
20世纪70年代开发的微胶囊包覆法可使有机颜料免受环境的影响,改变其性能,延长其发挥功效的时间,现广泛用于纺织、涂料、香料、油墨、塑料、橡胶、医药、化妆品等领域[1-7],近年来还因其具有特殊的应用性能,如光电导性、催化性能等,作为功能性有机颜料应用.与无机颜料相比,有机颜料具有色泽鲜艳,着色强度高、色谱品种繁多、生产过程比较简单,毒性低等特点,随着其应用领域的不断扩大,有机颜料产量大幅度增加,对颜料的各种性能要求也越来越高,需要更好的耐久性、耐冷热性、耐溶性及耐迁移性等.目前有关有机颜料的研究热点是颜料表面改性处理、颜料分散技术等[8-10].
本文在研究密胺树脂包覆耐晒黄G性能[11]的基础上,用界面聚合法(以PVP为囊壁)和原位聚合法[12-14](以UF为囊壁)制备了耐晒黄G颜料微胶囊,并对合成条件及胶囊的性能进行了分析测定.
1 实验仪器与试剂
聚乙烯吡咯烷酮、硫酸钠、硫酸铵、硫酸镁、硫酸钾、氯化钠、硝酸钠、尿素、甲醛、氢氧化钠、氯化铵、乙酸、柠檬酸、甲酸和丙酮均为化学纯,耐晒黄G为工业级.
2 制备原理与方法
2.1 制备原理
界面聚合法(以PVP为囊壁):用PVP微胶囊化有机颜料是通过引发单凝聚来实现的.在室温或冷却时,通过加入分离剂来引发分离,分离剂可以是具有很好的水亲合力的无机固体或者有机液体.一般是采用非水溶剂或者是无机盐类,使其中一相分离成粘滞液体,并能够沉积在颜料粒子表面,形成包覆的有机膜.有机颜料粒子分散于PVP溶液中,通过沉积来微胶囊化有机颜料.沉积过程所加的聚合物先分散于水中,然后沉积于颜料颗粒上,形成稳定分散的微胶囊粒子.
原位聚合法(以UF为囊壁):应用原位聚合法制备微胶囊的缩聚工艺,是由水溶性单体或预聚体,在酸或碱的催化下,分子间脱去小分子,缩聚形成交联网状结构的非水溶性聚合物,沉积到油溶性囊芯表而并包覆形成微胶囊.本实验中尿素氮原子上的活泼H原子很容易跟甲醛反应,生成羟甲基化合物,而羟甲基化合物易脱水形成线性预聚体,其反应式如(1)、(2)所示:
CO(NH2)2+2CH2O→CO(NHCH2OH)2
(1)
nCO(NH2OH)2→CH2NHCONHCH2+(n-1)H2O
(2)
上述线性预聚体是可溶于水的,在酸和碱的催化作用下能发生缩聚反应,预聚体分子间进一步脱去小分子形成交联立体网状结构的非水溶性缩聚物.
2.2 制备方法
PVP包覆:在有搅拌的情况下向三口烧瓶中加入一定量的耐晒黄G、分散剂和水,同时在另一容器将一定质量的聚乙烯吡咯烷酮溶解在水中.然后把两溶液混合,搅拌均匀.再在搅拌条件下按一定速率加入17%的无机盐溶液(分离剂),或者是无水丙酮.加完后,经过沉积、过滤、洗涤和干燥,得到PVP包覆的微胶囊化耐晒黄G.
UF包覆:取一定量的颜料,加入蒸馏水和适量的分散剂,高速搅拌分散;取30 g尿素与81 g(HCHO)含量为37%的甲醛(其物质的量之比为n尿素∶n甲醛=1∶2)混合,用NaOH调节pH=8.0,70 ℃下反应1 h得粘稠透明脲-甲醛预聚物,将其与分散液混合,充分搅拌使预聚物溶解于分散介质水中,分批加入催化剂NH4Cl,使介质pH值逐渐减小到3.5左右,加热至70 ℃,继续包覆并逐渐固化,当pH值降至1.5~2.0时加入100 mL热水,保温1 h,用NaOH调节pH=7.0,冷却后离心、干燥,得到UF包覆的颜料微胶囊.
3.1.4 部分较难测量液体出入量项目的漏记现象 出量错误调查中,不显性失水未记41.35%,痰液量未记37.83%,渗出未记9.68%,引流漏记10.85%,呕吐未记0.29%,说明对于那些需要进行换算或者较难收集的项目更易出错,如大小便失禁、痰液、汗液、呕吐物、伤口渗出液以及引流液的记录与计算是问题所在。皮肤、呼吸、气管切开等较难测量项目的漏记现象较为普遍。
3 结果与讨论
3.1 PVP包覆耐晒黄G
3.1.1 分离剂的选择 分离剂是PVP微胶囊化有机颜料的重要试剂.为了确定分离剂盐类的种类和用量,分别采用空白的PVP水溶液进行实验.分别在200 mL质量分数为4%的PVP水溶液中,加入几种质量分数均为17%无机盐,对比其开始沉淀和沉淀完全的用量,其结果如表1所示.

表1 分离剂的种类和用量关系
从表1可以看出,硫酸根离子引起的沉积效果较好,其原因是硫酸根离子所带的电荷比一价的离子大,对PVP的吸引力大.在不同的试验中可以选用不同的硫酸盐,考虑到操作过程应易于控制和成本等因素,在后续实验中选用用量适中的Na2SO4为分离剂.
3.1.2 分离剂添加方式对微胶囊粒径的影响 将颜料耐晒黄G分散于水中,同时将PVP分散于另一份水中,然后把两液混合,加入表面活性剂乙醇溶液.在高速搅拌的条件下按不同的方式加入质量分数为17%的硫酸钠溶液.考察添加速度对微胶囊形态的影响,结果如图1所示,按不同的方式加入分离剂,微胶囊粒径有较大的不同,加入速度慢形成的微胶囊粒径小,而且粒径分布比较集中.3种加入方式中以按20 mL/min的速度加入效果最好.这是因为微胶囊在形成的时候,随着分离剂的加入聚乙烯吡咯烷酮沉积在颜料离子表面,如果加入速度过快,沉积的速度也就快,壁材还有可能没有沉积到颜料粒子表面就已经聚集,这样微胶囊包膜就不会很致密,而且有可能有些粒子包膜过厚,而有些粒子却包覆不完全或者没有包覆.所以在试验中采用滴加的方法,速度为20 mL/min.
3.1.3 分散剂质量分数对微胶囊粒径的影响 对耐晒黄G进行包覆时,分散剂质量分数对微胶囊粒子的平均粒径有较大的影响,根据本课题组对包覆耐晒黄G中对分散剂效果的研究[11],在此选用分散剂3,其加入量对成型微胶囊平均料径的影响结果如图2所示,分散剂质量分数对粒径有很大的影响,随着分散剂质量分数的增加,成型微胶囊的平均粒径逐渐减小,当分散剂质量分数为2.5%时,微胶囊的平均粒径已小于2 μm,再增加分散剂的质量分数,效果不明显,所以选择加入分散剂的质量分数为2.5%.
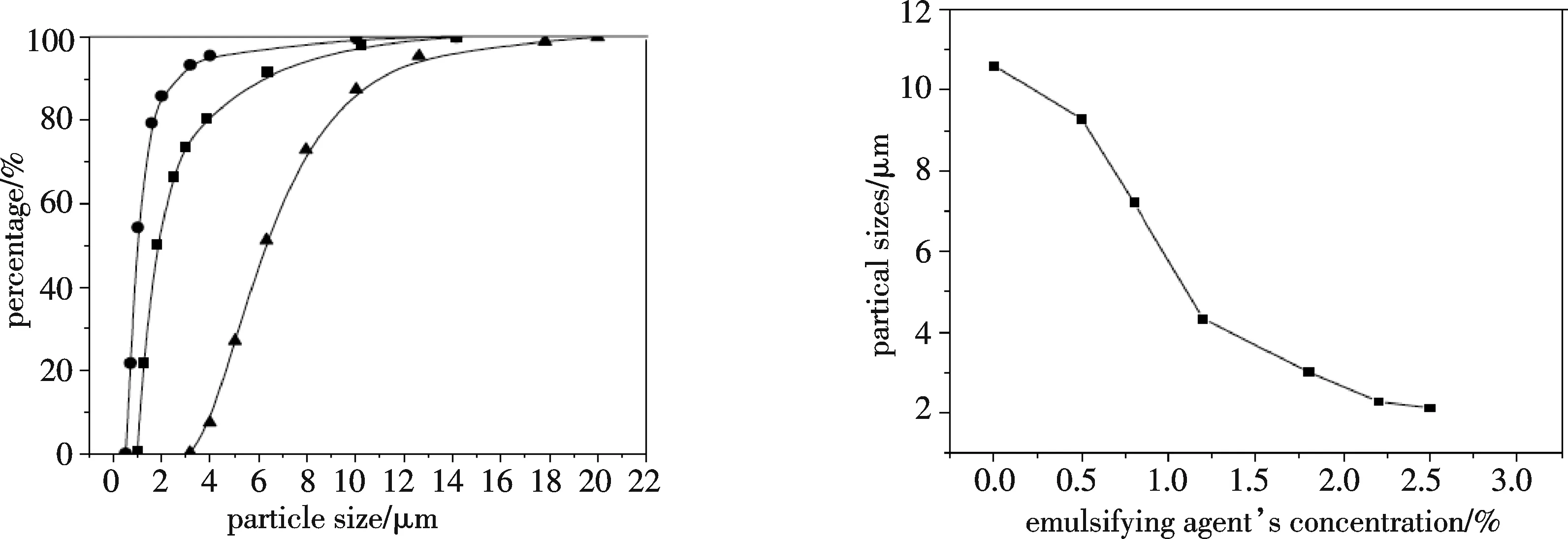
▲——一次加入;●——分3次加入;■——按20 mL/min加入 图1 Na2SO4添加速度对微胶囊形态分布的影响 图2 分散剂质量分数与微胶囊平均粒径的关系
3.1.4 囊材含量对微胶囊粒径的影响 囊材含量对微胶囊的粒径的影响主要是由于囊材含量的变化引起芯材/囊材比率的变化,从而导致微胶囊的粒径分布和成囊性能等方面变化,其结果如图3所示.在相同的乳化时间和搅拌速度下,当芯材的用量不变时,随着囊材含量的增加,芯材/囊材的比率随之减小,包覆在芯材上的囊材量也随之增加,从而导致微胶囊的粒径分布范围逐渐扩大,平均粒径也增加.而当囊材的含量小于15%时,成囊性能较差,会产生包覆不完全的现象.因此,选择包覆时囊材的含量为15%.
3.2 UF包覆耐晒黄G
3.2.1 反应介质pH值的影响
本实验中作为囊壁原料的脲-甲醛预聚物是由尿素和甲醛在碱性条件下,经加成反应生成的以二羟甲脲为主的水溶性羟甲基脲.它的分子中存在未反应的游离羟甲基、氨基、亚氨基等活性基团,在酸性条件下,易于进行缩聚反应,首先脱水形成以亚甲基键和少量醚键连接的线型或支链型低相对分子质量物,加热或加入固化剂后分子间交联形成体型网状结构.若在碱性条件下反应,则羟甲基之间不直接反应形成亚甲基键,而是形成二亚甲基醚键,再进一步分解放出甲醛形成亚甲基键,所以碱性介质使缩聚反应时的活性基团活性降低,反应速度相当慢.固化时使交联度下降,影响胶囊壁强度.因此选择在酸性条件下进行缩聚反应.实验表明,终点pH值越低,三维网状结构越趋紧密,形成的微胶囊越坚固,其结果如表2所示,控制终点pH值在1.5~2.0,形成的微胶囊结构紧密、坚固.
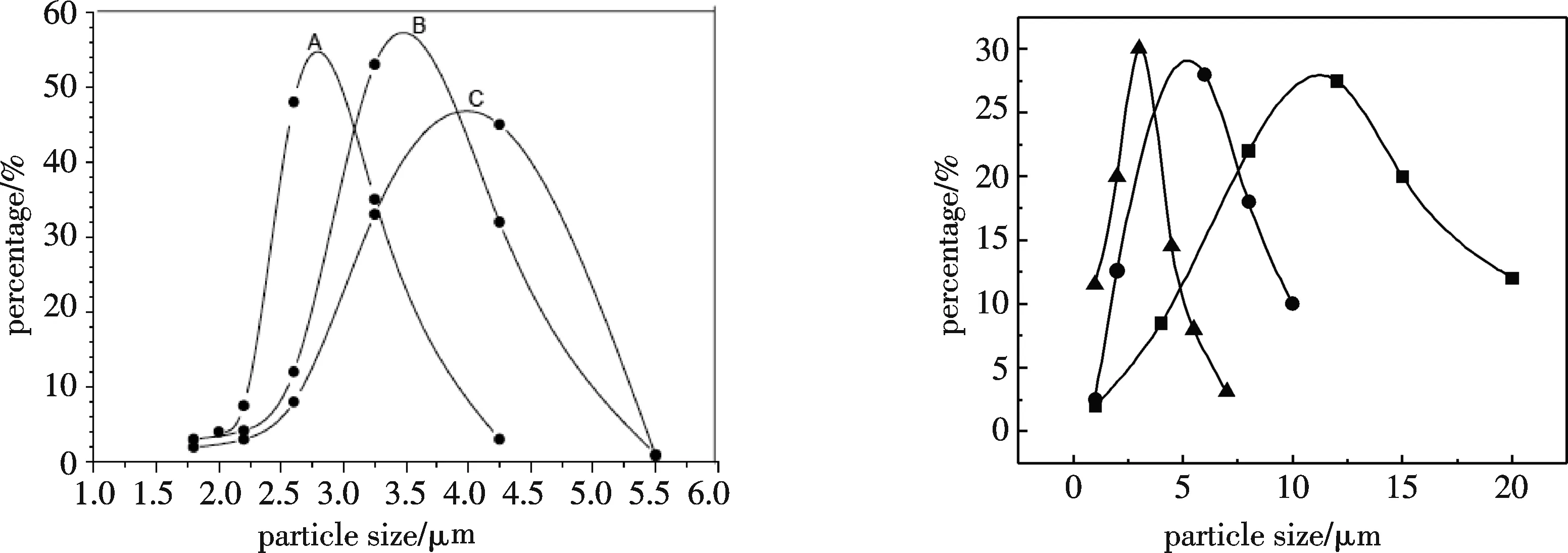
A——囊材含量15%; ■——酸化时间2 h,平均粒径d=11.4 μm B——囊材含量20%; ●——酸化时间3 h,平均粒径d=5.3 μm C——囊材含量30% ▲——酸化时间4 h,平均粒径d=3.2 μm 图3 囊材用量与微胶囊粒径的关系 图4 酸化时间对粒径及其分布的影响

序 号终点pH值微胶囊结构1>4.0表面结构松散,易破裂23.0表面结构松散32.0表面结构紧密41.5表面结构紧密,坚固
3.2.2 催化剂的影响 缩聚反应阶段常用的酸性催化剂有甲酸、乙酸、柠檬酸等价格低的有机酸.但用有机酸酸化不能使体系的终点pH值小于2.5,会影响微胶囊的结构.若用强酸弱碱盐NH4Cl做催化剂,在缩聚反应中能与水及游离甲醛反应放出强酸使pH值降低,其反应式如 (3)、(4)所示:

(3)
(4)
反应时加热有利于NH4Cl分解.氯化铵和有机酸比较,在相同反应条件下更易将体系调至较低pH值,结果见表3.NH4Cl还可以消耗过量的游离甲醛,并在反应中起到固化剂的作用,使囊壁更坚固,反应也比较缓和,容易控制.实验表明,选用价格低廉的工业氯化铵即可满足要求.

表3 不同催化剂对终点pH值的影响
3.2.3 酸化时间的影响 用显微镜监测缩聚反应过程,可观察到随着酸性催化剂的加入,pH值降低到5.0以下时囊壁开始形成.若pH值迅速降到2.5以下,则聚合急剧,囊壁形成太快,造成微胶囊粒径分布不匀;同时体系中的粘度增加太快,易结块,微胶囊的质量不易控制.实验发现,适当延长酸化时间,反应终点时pH值就低,得到微胶囊粒径小、粒径分布均匀、微胶囊越坚固,但酸化时间超过4 h后,微胶囊粒径分布基本不变.实验结果如图4所示:将酸性催化剂分批缓慢加入,控制终点pH值在1.5,酸化时间为4 h时,得到平均粒径为3.2 μm的微胶囊.
3.2.4 反应温度的影响 在其他反应条件相同时,反应温度对微胶囊的粒径影响如图5所示.随着反应温度的增加,形成的微胶囊平均粒径先保持不变,随后急剧上升.这是由于反应温度过高时,缩聚反应太快,导致形成的微胶囊平均粒径迅速增加;而在反应温度较低时,尽管形成的微胶囊粒径和反应温度为70 ℃时的微胶囊粒径相当,但是由于反应温度低时反应速度较慢,反应时间延长,形成的缩聚物相对分子量较小,致使微胶囊固化速度过慢甚至不固化,得到的微胶囊成品量很少,即使调至较低终点pH值,最终也只能得到少量成品微胶囊.而在实验过程中,先在低温下酸化,然后控制温度为70 ℃下反应包覆并逐渐固化,固化时间为2 h,停止反应后离心、干燥可得到球形、分散性良好、成品量较多的固体微胶囊,其电镜照片如图6所示,统计得到微胶囊平均粒径约为3.2 μm.

图5 反应温度对微胶囊平均粒径的影响 图6 包覆理想的微胶囊表面结构
4 结论
(1) 用界面聚合法制备了以PVP为囊壁的耐晒黄G颜料微胶囊,优化的包覆条件为:以Na2SO4为分离剂,按20 mL/min的速度加入;囊材含量为15%;分散剂用量为2.5%.可以制得包覆良好,粒径分布均匀且平均粒径小于4 μm的流动性固体微胶囊.
(2) 用原位聚合法在酸催化作用下制备了以UF为囊壁的耐晒黄G颜料微胶囊,优化的缩聚反应条件为:以NH4Cl为酸性催化剂,催化剂分批加入,制得的耐晒黄G微胶囊包覆良好,微胶囊紧密坚固,粒径分布均匀且平均粒径达3.2 μm.
参考文献:
[1] HONG K, PARK S. Melamine resin microcapsules containing fragrant oil synthesis and characterization[J]. Mater Chem Phys, 1999, 58(2): 128-131.
[2] 宋 健, 刘东志, 张天永. 微胶囊及微胶囊化技术的研究进展[J]. 化工进展, 1999(1): 42-44.
[3] 徐冬梅, 张可达, 王 平, 等. 微胶囊的功能及应用[J]. 精细石油化工, 2003(6): 55-59.
[4] 葛艳蕊, 冯 薇. 原位聚合法制备玫瑰香精微胶囊的研究[J]. 日用化学工业, 2003, 33(5): 337-339.
[5] 郭慧林, 李恒欣, 范 辉, 等. 原位聚合法制备红色电子墨水微胶囊[J]. 功能材料, 2006, 37(4): 559-561.
[6] PARK B J, LEE J Y, SUNG J H,etal. Microcapsules containing electrophoretic suspension of TiO2modified with poly(methyl methacrylate)[J]. Curr Appl Phys, 2006, 6(4): 632-635.
[7] SIMON B,CHRISTOPHER A,GEOFFRE M.An atomic force microscopy study of weathering of polyester/melamine paim surfaces[J]. Mater Chem Phys, 2001, 42(1-2): 49-58.
[8] 蔡 涛, 王 丹, 宋志祥, 等. 微胶囊的制备技术及其应用[J]. 化工中间体, 2009(12): 6-13.
[9] 冀林仙, 史晓滨. 助剂对微胶囊剂稳定性影响研究[J]. 现代农药, 2007, 6(5): 22-23.
[10] 邓 磊, 林休休, 刘小红, 等. Fe3O4聚脲微胶囊的制备及性能[J]. 苏州大学学报:自然科学版, 2007, 23(3): 70-74.
[11] 湛雪辉, 荀育军, 甘均良, 等. 影响密胺树脂包覆耐晒黄G的条件研究[J]. 湖南师范大学自然科学学报, 2007, 30(1): 60-63.
[12] 戴杜雁, 张万长, 张婉南, 等. 对原位聚合法制备微胶囊技术的研究[J]. 塑料工业, 1994(2): 27-31.
[13] 冀林仙, 石敏先, 郑保忠. 脲醛树脂微胶囊的制备[J]. 材料导报, 2005, 19(5): 109-110.
[14] JIMENEZ M, GARCIAH S, BERISTAIN C I. Spray-drying micro encapsulation and oxidative stability of conjugated linoleic acid[J]. Eur Food Res Technol, 2004, 219(6): 588-592.
