紫外分光光度法测定甘草中总黄酮的含量
2012-11-21贠亚波兀江波曹峡萍三门峡市中医院药剂科河南三门峡472000
高 宏,贠亚波,兀江波,曹峡萍(三门峡市中医院药剂科,河南三门峡 472000)
紫外分光光度法测定甘草中总黄酮的含量
高 宏*,贠亚波,兀江波,曹峡萍(三门峡市中医院药剂科,河南三门峡 472000)
目的:建立甘草总黄酮的含量测定方法。方法:以甘草苷为对照品,以10%KOH溶液为显色剂,优化显色时间、显色剂用量,并最终建立紫外分光光度法测定甘草总黄酮。结果:确定最佳的显色剂用量为1.5mL、显色时间为15min;甘草苷的进样浓度在0.012~0.030mg·mL-1范围内与吸光度呈良好的线性关系(r=0.9998),平均加样回收率为99.0%,RSD=2.60(n=9)。结论:该方法快速、简单、有效,适用于甘草总黄酮含量的测定。
甘草;总黄酮;含量测定;紫外分光光度法;甘草苷
甘草系豆科植物甘草Glycyrrhiza uralensia Fisch.、胀果甘草G.inflate Bat.或光果甘草G.glabra L.的干燥根及根茎[1]。甘草有效部位的提取一直是研究的热点,其有效成分主要以二氢黄酮类和查尔酮类化合物为主,在抗氧化、保肝、抗肿瘤方面有着很好的药理活性。对于其总黄酮的检测方法多以芦丁为对照品[2];若以柚皮苷[3]为对照品,因其不是甘草中的有效成分,所以检测结果存在较大差异。笔者尝试以甘草苷作为对照品,采用紫外分光光度法测定甘草中总黄酮的含量。
1 仪器与试药
8453型紫外-可见分光光度计(美国安捷伦公司);TDA-8002型电热恒温水浴锅(余姚市东方电工仪器厂);KQ300DA型数控超声仪(昆山市超声仪器有限公司,功率:300W,频率:53kHz)。
甘草苷对照品(中国食品药品检定研究院,批号:111610-200503);氢氧化钠、氢氧化钾、无水乙醇均为分析纯;甘草购于安徽亳州市永刚饮片厂有限公司(批号及产地:20110301,内蒙古阿拉善;20110415,宁夏盐池;20110510,新疆巴楚),粉碎,过100目筛,备用。
2 方法与结果
2.1 对照品溶液的制备
精密称定已干燥的甘草苷对照品适量,用70%乙醇溶解并定容于25mL量瓶中,制得浓度约为0.15mg·mL-1的对照品溶液。摇匀后冰箱中贮藏,备用。
2.2 供试品溶液的制备
取甘草粉末(批号:20110510)约0.2g,精密称定,置50mL量瓶中,加70%乙醇45mL,超声处理30min,放冷,加70%乙醇至刻度,摇匀,过滤,取续滤液,即得。
2.3 最大吸收峰的确定
精密吸取甘草苷对照品溶液1.0mL,加入10%KOH溶液1.0mL,用70%乙醇稀释至5mL,摇匀后室温放置5min。另取甘草苷对照品溶液1.0mL,置5mL量瓶中,用70%乙醇稀释至刻度,作为空白对照。2种溶液于190~500nm波长范围内扫描,结果在407nm波长处有最大吸收(见图1)。
精密吸取供试品溶液0.5mL,置5mL量瓶中,加入10%KOH溶液1.0mL,用70%乙醇稀释至5mL,摇匀后室温放置5min。再取供试品溶液0.5mL,直接用70%乙醇定容至5mL,作为空白对照。2种溶液于190~900nm波长范围内扫描,结果在407nm波长附近有最大吸收(见图1)。
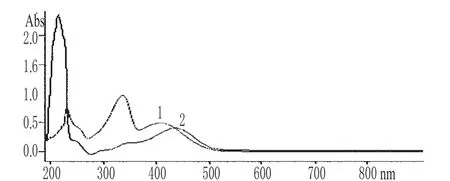
图1 紫外吸收光谱图1.甘草苷对照品;2.供试品Fig 1 UV absorption spectrum1.liquiritin control;2.test sample
综上,选取407nm为检测波长。
2.4 显色剂用量及显色时间的确定
精密吸取对照品溶液1mL,置于5mL量瓶中,分别加10%氢氧化钾溶液0.8、1.0、1.25、1.5mL,立刻加70%乙醇定容。另取对照品溶液1mL,加70%乙醇至5mL,作为空白对照。每隔2.5min在407nm波长处测定各溶液的吸光度。结果表明,加入10%氢氧化钾溶液1.5mL稀释后,放置时间15min,吸光度值趋于稳定,故本试验选择显色剂的用量为1.5mL、显色时间为15min。
2.5 线性关系考察
精密吸取甘草苷对照品溶液0.4、0.5、0.6、0.7、0.8、0.9、1.0mL,分别加入10%KOH溶液1.5mL,用70%乙醇稀释至5mL,室温放置15min,在407nm波长处测定吸光度。以吸光度(Y)为纵坐标,对照品的浓度(X,mg·mL-1)为横坐标,进行线性回归,得回归方程为Y=21.9974X-0.0055(r=0.9998,n=7)。结果表明,甘草苷的进样浓度在0.012~0.030mg·mL-1范围内与吸光度呈良好的线性关系。
2.6 重复性试验
精密吸取同一批(批号:20110510)供试品溶液6份,每份0.4mL,按“2.5”项下方法操作;再吸取同量供试品溶液,直接用70%乙醇定容至5mL,作为空白对照,于407nm波长处测定吸光度。结果,样品中总黄酮含量为2.79%,RSD=1.45%(n=6),表明本方法重复性良好,
2.7 精密度试验
精密吸取对照品溶液适量,按“2.5”项下方法制得显色溶液,取各显色溶液及其空白溶液于407nm波长处测定,共操作5次,以吸光度值计算。结果,RSD=1.1%(n=5),表明仪器精密度良好。
2.8 稳定性试验
取供试品溶液(批号:20110510)适量,分别于0、1、2、3、4、6、8h显色,按“2.5”项下方法测定吸光度。结果,RSD=1.46%(n=7),表明供试品溶液在8h内稳定性良好。
2.9 加样回收率试验
精密称取已知含量(2.79%)的甘草样品(批号:20110510)9份,每份0.1g,精密加入甘草苷对照品,按“2.2”项下方法制备供试品溶液,照“2.5”项下方法操作,计算加样回收率,结果见表1。

表1 加样回收率试验结果(n=9)Tab 1Results of recovery test(n=9)
2.10 样品含量测定
取不同批号甘草粉末各约0.2g,精密称定,置50mL量瓶中,加70%乙醇45mL,超声处理30min,放冷,加70%乙醇至刻度,摇匀,滤过。精密吸取滤液0.5mL,加入1.5mL 10%KOH溶液1.5mL,用70%乙醇稀释至5mL,室温放置15min,于407nm波长处测定吸光度,计算样品含量。结果,3批甘草药材(批号:20110301、20110415、20110510)中总黄酮的含量分别为6.08%、2.82%、2.79%(n=3)。
3 讨论
2010年版《中国药典》中甘草的含量测定方法为检测甘草苷和甘草酸的含量,对于其所含的黄酮类化合物并无检测方法。采用紫外分光光度(UV)法测定甘草中总黄酮多用芦丁作对照品[2]、亚硝酸钠-硝酸铝-氢氧化钠显色。但是,笔者对采用亚硝酸钠-硝酸铝-氢氧化钠显色后的甘草苷对照品溶液进行扫描,发现其最大吸收峰出现在400nm左右波长处;而用芦丁作对照品显色后测定的波长在500nm以上,产生误差较大,效果不理想。又有用柚皮苷[3]作对照品、氢氧化钾显色测定甘草中总黄酮含量的方法,但是甘草中可能没有柚皮苷,影响试验结果的准确性。还有用甘草苷作对照品、氢氧化钠显色的方法[4],利用甘草中黄酮类成分主要为甘草苷,而甘草苷为二氢黄酮类成分,遇到碱性试剂转变为查尔酮,显紫红色。但是试验结果证明用氢氧化钠作显色剂,碱性不如氢氧化钾强,所测吸光度浮动范围较大,不如氢氧化钾作显色剂稳定。经综合考虑,笔者选用甘草苷为对照品,以氢氧化钾作为显色剂测定甘草中总黄酮的含量,该方法稳定、简单、快速、有效。
样品药材中产自内蒙古的为野生甘草,其余的均为家种甘草,从结果看野生甘草中总黄酮的含量较高。2005年版《中国药典》规定甘草中甘草苷的含量不得少于1%[5],而2010年版《中国药典》规定甘草中甘草苷的含量不得少于0.5%。通过笔者对甘草质量的研究发现,若按照2005年版《中国药典》的规定,全国相当一部分甘草中甘草苷的含量达不到此要求;2010年版《中国药典》降低标准或为此原因。
[1] 国家药典委员会.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:80.
[2] 封士兰.甘草黄酮的提取分离和含量测定[J].兰州医学院学报,1998,24(4):20.
[3] 张雪辉,赵元芬,陈建民.甘草中总黄酮的含量测定[J].中国中药杂志,2001,26(11):746.
[4] 韩 博,陈 文,翟科峰,等.甘草苷作对照品测定乌拉尔甘草中总黄酮的含量[J].石河子大学学报(自然科学版),2006,24(4):472.
[5] 国家药典委员会.中华人民共和国药典(一部)[S].2005年版.北京:化学工业出版社,2005:60.
Content Determination of Total Flavonoids from Radix Et Rhizoma Glycyrrhizae by UV Method
GAO Hong,YUAN Ya-bo,WU Jiang-bo,CAO Xia-ping(Dept.of Pharmacy,Sanmenxia Hospital of TCM,Henan Sanmenxia 472000,China)
OBJECTIVE:To establish the method for the content determination of total flavonoids from Radix Et Rhizoma Glycyrrhizae.METHODS:Liquiritin and 10%KOH solution were used as substance control and color reagent to optimize color developing time and amount of color reagent.At last,UV spectrophotometry was established for the content determination of total flavonoids from Radix Et Rhizoma Glycyrrhizae.RESULTS:Optimal amount of color reagent was 1.5mL and color developing time was 15min;the linear range of liquiritin was 0.012~0.030mg·mL-1(r=0.9998)with an average recovery of 99.0%(RSD=2.60,n=9).CONCLUSION:The method is rapid,simple and effective,and it is suitable for the content determination of content determination of total flavonoids from Radix Et Rhizoma Glycyrrhizae.
Radix Et Rhizoma Glycyrrhizae;Total flavonoids;Content determination;UV spectrophotometry;Liquiritin
R284.1;R927.2
A
1001-0408(2012)07-0642-02
DOI 10.6039/j.issn.1001-0408.2012.07.24
*副主任药师。研究方向:药品质量鉴定,临床药学。电话:0398-2843570。E-mail:zyyyjk@163.com
2011-09-22
2011-12-27)
