HPLC法测定联苯乙酸凝胶含量和有关物质
2012-11-21汪平张军兵武惠斌聂红梅河南羚锐制药股份有限公司河南新县465550北京羚锐伟业科技有限公司北京100070
汪平,张军兵,武惠斌,聂红梅(1.河南羚锐制药股份有限公司,河南新县465550;2.北京羚锐伟业科技有限公司,北京 100070)
HPLC法测定联苯乙酸凝胶含量和有关物质
汪平1,2*,张军兵1,2,武惠斌1,2,聂红梅1,2(1.河南羚锐制药股份有限公司,河南新县465550;2.北京羚锐伟业科技有限公司,北京 100070)
目的:建立测定联苯乙酸凝胶的含量和有关物质的方法。方法:采用高效液相色谱法。色谱柱为Kromasil C18,流动相为甲醇-0.1%冰醋酸(65∶35),流速为1.0mL·min-1,检测波长为254nm。结果:联苯乙酸检测浓度线性范围为0.3~60μg·mL-1(r=0.9998),低、中、高浓度平均回收率分别为100.3%、101.2%、99.8%,RSD=0.24%(n=9);3批样品主药和有关物质含量均符合限度要求。结论:本方法简便快速、重复性好,可用于控制联苯乙酸凝胶的质量。
联苯乙酸凝胶;有关物质;含量;高效液相色谱法
联苯乙酸为非甾体抗炎药,由日本Lederle公司研制,其凝胶剂1986年首先在日本上市,局部用于消炎镇痛,主要用于变形性关节炎、肩周炎、腱鞘炎、腱周炎、肌肉痛、外伤后肿胀疼痛、软组织损伤等的镇痛消炎[1]。为控制该制剂质量、保证临床疗效,笔者对其含量和有关物质测定方法进行了研究。
1 仪器与试药
LC-20A型高效液相色谱(HPLC)仪,包括SIL-20A自动进样器、CTO-20A柱温箱、SPD-20A紫外-可见检测器、LC Solution色谱工作站(日本岛津公司);CP225D型电子天平(德国Sartorius公司)。
样品:联苯乙酸凝胶(北京羚锐伟业科技有限公司,批号:000703、000704、000705,规格:10g∶0.3g);联苯乙酸原料药(天津药物研究院,批号:000512,纯度:99.98%);联苯乙酸对照品(英国LGC公司,批号:Y0000731,纯度:100%);联苯对照品(中国食品药品检定研究院,批号:110843-9501,纯度:100%);邻苯基苯甲酸对照品(批号:A0270674,纯度:99.8%)、4-乙酰联苯对照品(批号:MKBG0866V,纯度:99.9%)均系美国SigmaAldrich公司产品;甲醇为色谱纯,其他试剂均为分析纯。
2 方法与结果
2.1 有关物质
邻苯基苯甲酸为联苯乙酸合成过程中产生的副产物,联苯为联苯乙酸的起始原料,4-乙酰联苯为联苯乙酸的降解产物。但因为邻苯基苯甲酸在联苯乙酸原料药的质量研究中并未检出,故在联苯乙酸凝胶中不作为已知杂质考察,但将其与联苯乙酸的分离度用以考察系统适用性。
2.1.1 色谱条件[2]。色谱柱:Kromasil C18(250mm×4.6mm,5μm);柱温:30℃;流动相:甲醇-0.1%冰醋酸(65∶35),流速:1.0mL·min-1;检测波长:254nm。理论板数按联苯乙酸峰计不低于5000。
2.1.2 测定法。供试液:取样品适量(相当于联苯乙酸3mg),精密称定,加流动相溶解并定量稀释至10mL量瓶中,摇匀,过滤,即得。对照液(1):取供试液3倍体积,加流动相稀释到100倍体积,再取此溶液1倍体积加流动相稀释到10倍体积,摇匀,即得。对照液(2):取4-乙酰基联苯与联苯对照品适量,精密称定,以少量甲醇溶解后,加流动相定量稀释成每1mL中含4-乙酰联苯0.3μg、联苯0.3μg的混合溶液,即得。
取对照液(1)20μL进样,调节检测灵敏度,使主成分峰高约为满量程的15%,再取供试液和对照液(1)、(2)各20μL,分别进样,记录色谱图至主成分峰保留时间的3倍。供试液的色谱图中如出现与对照液(2)中2个主峰相对应的峰,其任何一个峰面积均不得大于相对应的主峰面积(每个0.1%);其他单个杂质峰面积均不得大于对照液(1)主峰面积的1/3(0.1%);未知杂质总和不得过1.0%。空白基质、对照液(1)、对照液(2)、供试液色谱详见图1。
2.1.3 线性关系考察。取样品适量,照供试液的制备方法制成每1mL中约0.5mg的溶液,分别精密量取该溶液适量,加流动相制成每1mL中约含联苯乙酸2、4、6、8、10µg的溶液,分别进样。以浓度(c)为横坐标,峰面积(A)为纵坐标,拟合曲线,得方程A=5502.3c+45.1(r=0.9996)。结果表明,联苯乙酸检测浓度线性范围为2~10μg·mL-1。
2.1.4 重复性试验。取样品(批号:000703)6份,制备成供试液后测定,结果RSD=0.22%,表明本方法重复性良好。
2.1.5 强制降解试验。取联苯乙酸凝胶适量照供试液制备方法制备成1.5mg·mL-1的溶液作为贮备液。分别取贮备液适量用1mol·L-1的盐酸、1mol·L-1的氢氧化钠、80℃高温、30%双氧水、4000lx光照破坏放置3h后定容至浓度为0.3mg·mL-1的溶液,滤过,作为供试品溶液测定,并同法制备空白溶液测定。结果本品比较稳定,碱、高温条件下略有降解,约降解0.5%和0.8%,其他条件下几乎未降解,详见图2。
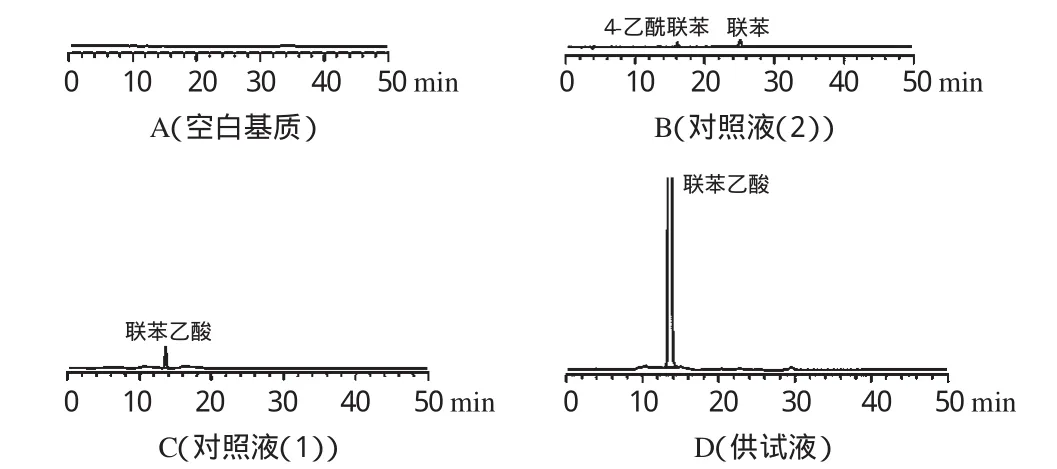
图1 有关物质检查中相关高效液相色谱图Fig 1 HPLC chromatograms of related substances

图2 有关物质降解试验高效液相色谱图1.碱破坏后样品;2.样品;3.酸破坏后样品;4.高温破坏后样品;5.空白;6.氧化破坏后样品;7.光照破坏后样品Fig 2HPLC chromatograms of degradation experiment of related substances1.sample destroyed by alkaline;2.sample;3.sample destroyed by acid;4.sample destroyed by high temperature;5.blank control;6.sample destroyed by oxidation;7.sample destroyed by light
2.1.6 稳定性试验。取供试液1份,分别放置0、2、4、6、8h测定,结果RSD=0.83%,表明供试液放置8h内稳定。
2.1.7 有关物质测定结果。3批样品中杂质含量均符合限度要求,详见表1。
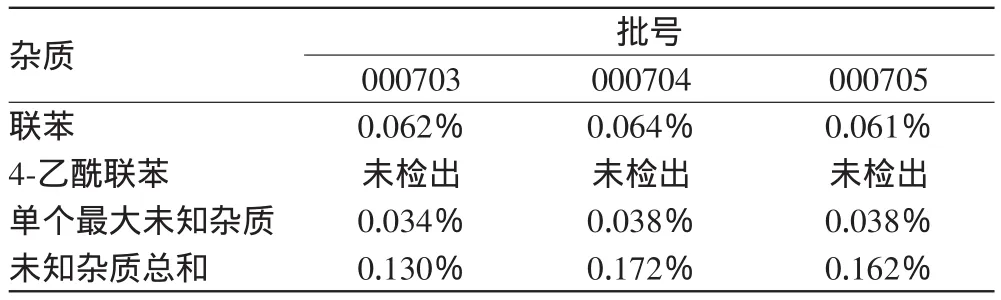
表1 3批样品中有关物质测定结果Tab 1 Result of related substances test of 3batches of samples
2.2 主药含量测定
2.2.1 色谱条件与线性关系考察。色谱条件同“2.1.1”项,线性关系考察方法操作同“2.1.3”项,分别制备浓度为0.3、3、6、9、12、24、30、36、47.5、60μg·mL-1的联苯乙酸对照品溶液进样分析,得回归方程A=151604c-25888(r=0.9998),联苯乙酸检测浓度线性范围为0.3~60μg·mL-1。
2.2.2 分离度测定。精密称取联苯乙酸和邻苯基苯甲酸对照品适量,用流动相制备成各含4μg·mL-1混合溶液,进样分析,记录色谱峰,计算2个主峰之间的分离度。结果,联苯乙酸和邻苯基苯甲酸保留时间分别为10.175、13.628min,分离度为7.93,详见图3。
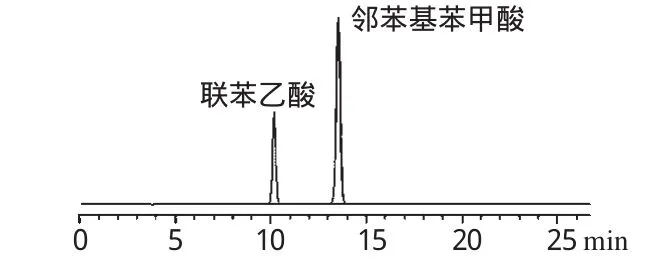
图3 分离度试验高效液相色谱图Fig 3 HPLC chromatograms of separation degree test
2.2.3 精密度试验。分别取浓度为3、6μg·mL-1的联苯乙酸对照品溶液,重复进样6次,结果RSD分别为0.65%和0.71%,表明本法精密度良好。
2.2.4 回收率试验。精密称取本品空白基质0.134g 9份,各置于50mL量瓶中,分别加入浓度为1mg·mL-1的联苯乙酸对照品溶液3(80%)、4(100%)、5(120%)mL各3份,加流动相定容至刻度摇匀;再精密量取此液1mL置于20mL量瓶中,加流动相稀释至刻度摇匀,滤过,进样测定。得低、中、高浓度平均回收率分别为100.3%、101.2%、99.8%,RSD=0.24%(n=9)。
2.2.5 重复性试验。取同一批样品(批号:000703)按“2.2.6”项下方法制备成供试品溶液,测定6次,结果RSD=0.45%,表明本方法的重复性好。
2.2.6 含量测定。精密称取样品适量(约相当于联苯乙酸4mg),加流动相溶解并定量稀释至100mL量瓶中,摇匀;精密量取5mL,置于50mL量瓶中,用流动相稀释至刻度,摇匀,过滤,取续滤液作为供试品溶液;取联苯乙酸对照品适量,加流动相溶解制备成4μg·mL-1溶液,作为对照品溶液;另取样品和邻苯基苯甲酸对照品适量,加流动相制备成浓度均为4μg·mL-1的混合溶液,作为分离度测定溶液。取分离度测定溶液适量进样,记录色谱峰,计算2个主峰之间的分离度,分离度不得小于3,否则试验无效。精密量取对照品溶液和供试品溶液各20μL进样,记录色谱峰,按外标法以峰面积计算含量,结果3批样品平均含量分别为100.30%、98.32%、100.40%。
3 论
联苯乙酸对照品溶液在254nm波长处有最大吸收,且空白基质在此处无干扰,故选254nm为其含量测定波长。
因本品结构简单,故选择通用的反相高效液相色谱法检测有关物质和含量。流动相中的冰醋酸主要起缓冲和改善分离的作用,既可提高方法的重复性,又可以改善峰形和分离情况,亦可防止联苯乙酸在分离过程中的解离和降解。
笔者曾经尝试过多种品牌的、不同长度的十八烷基硅烷键合硅胶色谱柱进行试验,结果分析效果均比较好。但是系统适用性试验仍然是非常必要的,如果邻苯基苯甲酸与联苯乙酸的分离度不足3,则很有可能导致有关物质测定时未知杂质之间的分离度小于1.5。
经过详细的方法学验证,结果表明,本方法简便快速、重复性好,可用于控制联苯乙酸凝胶的质量。
[1] 佚 名.联苯乙酸凝胶剂、搽剂(3.1类)[J].中国医药技术与市场,2006,6(2):46.
[2] 刘亚楠,王 伟.高效液相色谱法测定联苯乙酸原料及其制剂的有关物质[J].华夏医学,2010,23(4):375.
Content Determination of Felbinac Gel and Related Substances by HPLC
WANG Ping,ZHANG Jun-bing,WU Hui-bin,NIE Hong-mei(Henan Lingrui Pharmaceutical Co.,Ltd.,Henan Xinxian 465550,China)
WANG Ping,ZHANG Jun-bing,WU Hui-bin,NIE Hong-mei(Beijing Lingrui Weiye Technology Co.,Ltd.,Beijing 100070,China)
OBJECTIVE:To determine the contents of Felbinac gel and related substance.METHODS:HPLC method was adopted.The determination was performed on Kromasil C18with mobile phase consisted of methanol-0.1%glacial acetic acid(65∶35)at the flow rate of 1.0mL·min-1.The detection wavelength was set at 254nm.RESULTS:The linear range of felbinac was 0.3~60μg·mL-1(r=0.9998),and average recoveries were 100.3%,101.2%,99.8%at low,medium and high concentrations(RSD=0.24%,n=9).The contents of main component and impurity in 3batches of samples were in line with the requirements of limit.CONCLUSION:The method is simple,rapid and reproducible,and it can be used for the quality control of Felbinac gel.
Felbinac gel;Related substances;Content;HPLC
R927.2;R971+.1
A
1001-0408(2012)33-3142-02
DOI10.6039/j.issn.1001-0408.2012.33.28
*主管药师。研究方向:药理学。电话:010-82614159。E-mail:funnyping@sina.com
2012-01-18
2012-05-20)
