UPLC法与HPLC法测定加替沙星微球有关物质及微球的光稳定性评价Δ
2012-11-21林华庆张肖玲广东药学院药物研究所广东省药物新剂型重点实验室广州510006
邓 红,张 蜀,林华庆,张肖玲,田 璐(广东药学院药物研究所/广东省药物新剂型重点实验室,广州510006)
UPLC法与HPLC法测定加替沙星微球有关物质及微球的光稳定性评价Δ
邓 红*,张 蜀#,林华庆,张肖玲,田 璐(广东药学院药物研究所/广东省药物新剂型重点实验室,广州510006)
目的:采用超高效液相色谱法(UPLC)与高效液相色谱法(HPLC)测定加替沙星微球的有关物质,并进行微球的光稳定性评价。方法:UPLC法、HPLC法的色谱柱分别为Waters ACQUITY uplc®HSS T3、Diamosil C18(2),流速分别为0.6、1.0mL·min-1,进样量分别为5、50μL,流动相均为三乙胺磷酸溶液(pH 4.3)-乙腈(88∶12),检测波长均为293nm;另取微球和原料药在温度为25℃、湿度为60%、光照强度为6600lx下放置5、10d后,采用UPLC法考察杂质谱变化情况。结果:UPLC法和HPLC法检测加替沙星的最低检测限分别为0.001、4.1ng,最低定量限分别为0.002、15.3ng,检测时间分别为8、40min左右;微球和原料药经光照后杂质谱相似,但原料药中的部分杂质含量明显增加。结论:在拟定的色谱条件下,与HPLC法比较,UPLC法更快速、灵敏度更高;加替沙星制成微球后对光的稳定性增加。
超高效液相色谱法;高效液相色谱法;加替沙星微球;有关物质;光稳定性
加替沙星为第4代喹诺酮类抗菌药,其抗菌谱广,对G+细菌、衣原体、支原体等都有强大的杀菌作用,且光毒性较弱,对上述病原体引起的各种感染治疗均有效[1]。本课题研制的加替沙星微球将进一步制成关节腔埋植剂,用于治疗骨髓炎。迄今为止,加替沙星及其制剂的质量标准均未在各国药典收载,在药品质量标准中,对有关物质的控制是质量控制的重要环节,是稳定性考察的重要指标,同时也是药品评价的重要参数。超高效液相色谱(UPLC)是近年来发展迅速的基于小颗粒填料的液相色谱技术,相对于高效液相色谱(HPLC)法,能显著改善色谱峰的分离度和检测灵敏度,同时大大缩短分析周期[2,3],因此适用于微量复杂混合物如有关物质的分析。本文比较采用UPLC与HPLC法检测加替沙星微球的有关物质,并比较加替沙星原料药和微球对光的稳定性,为其埋植剂的开发提供参考,同时为加替沙星有关物质提供新的检测方法。
1 仪器与试药
UPLC H-Class液相色谱仪(美国Waters公司);Ultimate 3000液相色谱仪(美国Dionex公司)。
加替沙星原料药及对照品(上虞京新药业有限公司,批号:0703301,纯度:99.5%);加替沙星微球(自制,批号:20110901、20110902、20110903,含量分别为:4.48%、4.32%、4.60%);乙腈为色谱纯,水为重蒸馏水,三乙胺和磷酸均为分析纯。
2 方法与结果
2.1 加替沙星微球的制备
按处方量称取加替沙星和聚乳酸-羟基乙酸共聚物(PLGA,50∶50)(1∶12)置锥瓶中,加乙腈溶解得药液。另取含0.25%司盘80(W/V)的液状石蜡置具塞锥瓶中,800r·min-1搅拌下缓慢加入药液,密塞;搅拌10min后400r·min-1继续搅拌1h,缓慢滴入正己烷;3h后停止搅拌,用少量正己烷洗涤3次,干燥,即得。
2.2 色谱条件
HPLC和UPLC法均以三乙胺磷酸溶液(三乙胺溶液(1→100),用稀磷酸调pH(4.3±0.05))-乙腈(88∶12)为流动相,检测波长为293nm,柱温为室温;HPLC法色谱柱采用Diamonsil-C18(2)(250mm×4.6mm,5μm),流速为1.0mL·min-1,进样量为50μL;UPLC法色谱柱采用Waters ACQUITY uplc®HSS T3(50mm×2.1mm,1.8μm),流速为0.6mL·min-1,进样量为5μL。
2.3 溶液的制备
2.3.1 微球供试品溶液。取加替沙星微球适量(约相当于加替沙星10mg),研细,精密称定,置于50mL量瓶中,加乙腈6mL,超声振摇使溶解,再加三乙胺磷酸溶液稀释至刻度,摇匀,滤过,取续滤液,即得。
2.3.2 微球自身对照溶液。取上述供试品溶液1mL,置于100mL量瓶中,加流动相稀释至刻度,即得。
2.3.3 原料药供试品溶液。取加替沙星原料药约10mg,精密称定,置于50mL量瓶中,加流动相溶解并稀释至刻度,即得。2.3.4阴性对照溶液。按处方工艺制备空白微球,精密称取适量,依照“2.3.1”项下方法制备即得。
2.4 专属性试验
2.4.1 辅料干扰试验。取微球供试品溶液、原料药供试品溶液和阴性对照溶液各50μL注入HPLC仪,各5μL注入UPLC仪,记录色谱图,结果空白辅料不干扰加替沙星及其杂质的测定。HPLC和UPLC法检测时间分别为40、8min左右,见图1。
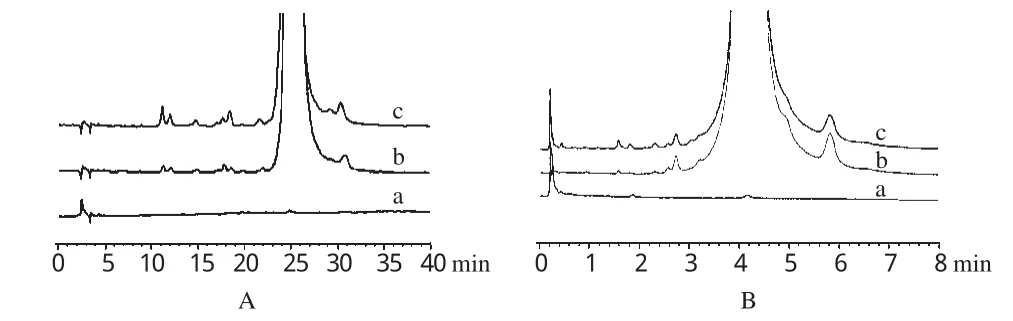
图1 干扰试验色谱图A.HPLC法;B.UPLC法;a.阴性对照溶液;b.原料药供试品溶液;c.微球供试品溶液Fig 1 HPLC and UPLC chromatograms of interfering test A.HPLC method;B.UPLC method;a.negative control solution;b.test sample solution of raw material;c.test sample solution of microspheres
2.4.2 强制降解试验。取微球供试品溶液、原料药供试品溶液和阴性对照溶液进行光照(紫外灯照射约48h)、高温(沸水浴加热2h,加乙腈补足减失体积)降解试验;取微球、原料药(约相当于加替沙星10mg)和空白微球适量,各3份,置50mL量瓶中,进行酸(加1mol·L-1盐酸溶液1mL)、碱(加3mol·L-1NaOH溶液1mL)、氧化(加3%H2O2溶液1mL)降解试验,再分别加乙腈6mL,超声振摇使溶解,48h后加三乙胺磷酸溶液稀释至刻度。取以上各溶液,滤过,取续滤液,采用2种方法进行分析。结果空白微球对上述5种破坏方法均较稳定;原料药和微球对酸、碱破坏较稳定,光照破坏下检出较多杂质。二者杂质谱相似,UPLC法对杂质的检测能力较强,微球降解试验的色谱图详见图2(其他图略)。
2.5 最低检测限和最低定量限试验
2.5.1 HPLC法。取原料药,用流动相稀释制成浓度分别为0.08176、0.3066μg·mL-1的溶液,进样分析,记录色谱图。以信/噪为3计算最低检测限为4.1ng(0.04%),以信/噪为10计算最低定量限为15.3ng(0.15%)。
2.5.2 UPLC法。取原料药,用流动相稀释制成浓度分别为0.2044、0.3066ng·mL-1的溶液,进样分析,记录色谱图。以信/噪为3计算最低检测限为0.001ng(0.0001%),以信/噪为10计算最低定量限为0.002ng(0.0002%)。
2.6 供试品溶液的稳定性考察
分别取原料药、微球的供试品溶液注入UPLC仪,于0、1、2、6、8h测定,记录峰面积,结果原料药和微球中杂质1、2、6峰面积在2h后均增加,因此溶液制备后应在2h内测定。
2.7 样品测定结果比较
取微球3批,依“2.3”项下方法制备成供试品溶液和自身对照溶液,按上述HPLC法和UPLC法测定。供试品溶液如显杂质峰,单个杂质峰面积不得超过自身对照溶液的主峰面积的0.5倍(0.5%),各杂质峰面积的和不得大于自身对照溶液主峰面积(1.0%),测定结果见表1。

表1 2种方法检测样品中有关物质的结果比较Tab 1 Comparison of determination results of related substances by 2methods
2.8 样品光稳定性的评价
取微球、原料药和空白微球(阴性对照)适量,置于洁净玻璃平皿中,放入智能型人工气候箱,设置温度为25℃、湿度为60%、光照强度为6600lx,于第5、10天时取样,按UPLC方法进行杂质谱分析,比较微球和原料药在光照射后的杂质谱变化,结果见图3。
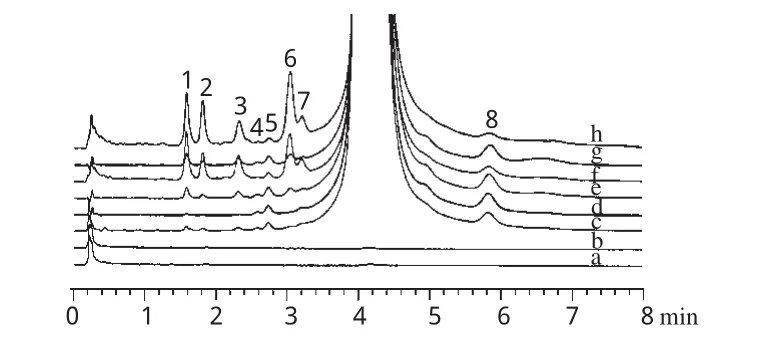
图3 加替沙星原料药/微球光降解试验杂质色谱图a.未光照阴性对照;b.光照10d阴性对照;c.未光照微球;d.未光照原料药;e.光照5d微球;f.光照5d原料药;g.光照10d微球;h.光照10d原料药;1~8.未知杂质Fig 3 Impurity chromatograms of gatifloxacin raw material and microspheres in light degradation testa.untreated negative control;b.negative control treated by light for 10d;c.untreated microspheres;d.untreated raw material;e.microspherestreated by light for 5d;f.raw material treated by light for 5d;g.microspheres treated by light for 10d;h.raw material treated by light for 10d;1~8.unknown impurities
由图3可见,微球和原料药经光照试验后,杂质谱相似,其中杂质4、5、8的峰面积基本没有改变,原料药的杂质1、2、3、6峰面积明显增加,而微球相对较稳定。据此,可认为将加替沙星制成微球后,对光的稳定性提高,杂质1、2、3、6是与加替沙星光照稳定性密切相关的主要杂质。为此以杂质1、2、3、6和总杂质含量为指标,进一步比较加替沙星原料药/微球光降解前后的杂质含量变化,结果表明微球处方工艺合理,详见图4。
3 讨论
(1)检测波长的确定。加替沙星在293nm波长有最大吸收[4],本试验HPLC法和UPLC法均与二极管阵列检测器(DAD)联用,通过全波长扫描,在293nm波长处主峰和杂质峰的紫外吸收均较强,说明可检出杂质的最大吸收也在293nm附近,故有关物质的检测波长定在293nm。

图4 加替沙星原料药/微球光照前后杂质含量变化比较Fig 4 Comparison of the contents of impurity in gatifloxacin raw material and microspheres before and after light
(2)溶液制备方法的选择。本品主要以聚乳酸为辅料制成微球,由于加替沙星和聚乳酸均溶于乙腈,因此先用乙腈溶解微球,保证主药完全释出;考虑到与流动相的一致性,再按流动相比例加入pH为4.3的三乙胺溶液(1→100)。
(3)大多数喹诺酮药物对酸、碱的稳定性较好,对光照稳定性差异较大。喹诺酮药物的光稳定性不仅与药物结构有关,且受处方工艺的影响。因此,对自制微球和原料药光照稳定性进行了比较,结果微球和原料药的杂质谱相似,说明杂质主要来自原料药。但相对于原料药,加替沙星制成微球后,对光的稳定性提高。
(4)本试验比较了2种方法测定加替沙星微球的有关物质,与HPLC法相比,UPLC法具有检测灵敏度更高、分析更快速、对杂质的检测能力更强等优势,在保证杂质检出能力的前提下,分析效率更高,分析时间更短,溶剂损耗更低,降低了分析成本。因此,UPLC法特别适合药物微量、复杂体系如有关物质的分析,值得推广应用。
[1] 杜 唯,刘护鱼.新一代氟喹诺酮类药物——加替沙星[J].西北药学杂志,2004,8(19):189.
[2] 郝桂明,唐素芳.超高效液相色谱在药物分析中的应用[J].天津药学,2009,21(6):64.
[3] 孙全乐,蔡广知,贡济宇.超高效液相色谱法测定人参中人参皂苷Re的含量[J].中国药房,2012,23(3):258.
[4] 邓 红,梁健健,张 蜀,等.HPLC法测定加替沙星聚乳酸纳米粒的含量[J].广东药学院学报,2008,24(3):236.
Evaluation of Light Stability of Gatifloxacin Microsphere and Related Substances by UPLC and HPLC
DENG Hong,ZHANG Shu,LIN Hua-qing,ZHANG Xiao-ling,TIAN Lu(Institute of Materia Medica of
Guangdong Pharmaceutical University/Guangdong Provincial Key Laboratory of Advanced Drug Delivery,Guangzhou 510006,China)
OBJECTIVE:To detect the related substances of Gatifloxacin microsphere by UPLC and HPLC,and to evaluate the light stability of microspheres.METHODS:UPLC was performed on Waters ACQUITY uplc®HSS T3column with the flow rate of 0.6mL·min-1and the injection volume was 5μL;HPLC was performed on Diamosil C18(2)column with the flow rate of 1.0mL·min-1and the injection volume was 50μL.The mobile phase was triethylamine-phosphoric acid solution(pH4.3)-acetonitrile(88∶12),and detection wavelength was set at 293nm.The chromatograms change of impurity was detected by UPLC after Gatifloxacin microsphere and raw material were kept at 25℃with moisture of 60%and light intensity of 6600lx for 5days and 10days.RESULTS:LOQ of gatifloxacin were 0.001ng and 4.1ng,and LOD were 0.002ng and 15.3ng for UPLC and HPLC.The determination duration were 8and 40min.The impurity chromatograms of microspheres and raw material were similar and the contents of some impurity in raw material increased.CONCLUSIONS:Under fixed chromatogram condition,UPLC method is proved to be more rapid and sensitive than HPLC method.The light stability of gatifloxacin is improved after being made into microspheres.
UPLC;HPLC;Gatifloxacin microspheres;Related substances;Light stability
R927.2
A
1001-0408(2012)33-3127-03
DOI10.6039/j.issn.1001-0408.2012.33.21
Δ广东省医学科研基金项目(A2010296)
*高级工程师。研究方向:药物新剂型和质量标准。电话:020-39352507。E-mail:dengh361@sohu.com
#通讯作者:教授,硕士。研究方向:药物新剂型。电话:020-39352507。E-mail:linzhangshu@tom.com
2012-03-20
2012-05-09)
