FCC过程中噻吩类硫化物转化规律的研究进展
2012-11-09吴群英达志坚朱玉霞
吴群英,达志坚,朱玉霞
(中国石化 石油化工科学研究院,北京 100083)
FCC过程中噻吩类硫化物转化规律的研究进展
吴群英,达志坚,朱玉霞
(中国石化 石油化工科学研究院,北京 100083)
综述了FCC过程中噻吩类硫化物的裂化脱硫机理和转化途径,并从转化率和选择性出发,分析了不同结构的噻吩类硫化物的反应特点。在FCC条件下,噻吩的转化率较低,而带有烷基侧链的噻吩和苯并噻吩均具有较高的转化活性,其中短侧链的烷基噻吩类硫化物易于发生异构化和脱烷基反应,而长侧链的烷基噻吩类硫化物易于发生侧链裂化和环化反应;反应体系中的其他烃类及催化剂的性质也对噻吩类硫化物的反应路径和转化率有一定的影响,其中大分子烷烃和环烷烃等供氢剂和氢转移活性高的催化剂均有利于噻吩类硫化物的裂化脱硫。在此基础上,进一步总结了典型的噻吩类硫化物的转化网络。
噻吩;苯并噻吩;流化催化裂化;氢转移
随着石油资源的重质化和劣质化以及进口原油中硫含量的增加,世界各国都将面临加工含硫或高硫原油的挑战,尤其是FCC工艺,加工一定数量的重质含硫原油将是炼油工业面临的共同趋势。FCC原料中的含硫化合物包括硫醇、硫醚、二硫化物和噻吩类硫化物,以噻吩类硫化物为主[1],比如典型的减压馏分油和渣油中的噻吩类硫约占总硫量的70%(w),而焦化馏分油和渣油加氢生成油中的噻吩类硫约占总硫量的80%(w)和85%(w)以上。
与此同时,环保法规也日趋严格,因此国内外很多学者对汽油和柴油的硫类型及含量分布进行了详细分析[2-3]。FCC汽油中噻吩类硫约占总硫量的80%(w),并主要以烷基噻吩为主,而硫醇、硫醚和二硫化物所占比例很低。Corma等[4]发现,在FCC过程中,噻吩、苯并噻吩和二苯并噻吩等具有稳定的芳环结构,很难发生裂化脱硫,而活性硫化物是很容易分解成H2S而脱除的。Dupain等[5]也认为很难通过改变FCC工艺参数(如温度、剂油比和停留时间等)来促使噻吩类硫化物裂化生成H2S或进入价值较低的重循环油中。由此可见,对噻吩类硫化物转化机理和规律的深入研究具有非常重要的实际意义和社会价值。由于原料类型、催化剂性质和反应条件的差异,研究者对噻吩类硫化物在FCC过程的转化机理持有不同的观点[6-8]。
本文主要对噻吩类硫化物的转化机理和路径的研究进展进行了综述,并讨论了不同结构的噻吩类硫化物的反应特点,以及反应体系中其他烃类和催化剂的性质对噻吩类硫化物转化路径的影响;在此基础上,进一步总结了重油体系中典型的噻吩类硫化物的转化网络。
1 噻吩类硫化物的转化机理和路径
噻吩结构简单,并具有一定的代表性,因而常作为研究噻吩类硫化物转化机理的模型分子。噻吩分子上的硫原子有两对孤对电子,其中一对处在6电子π键系统,另一对则与噻吩环共平面,处于独立状态,这种电子云排布使得噻吩具有芳环的性质,结构稳定,一般难以直接开环裂化。
众多研究表明[7-9],含硫化合物在酸性催化剂上的反应机理与FCC过程中烃类的裂化反应相似[10],都遵循碳正离子反应机理。由于噻吩环上的硫原子具有给电子的共轭效应,提高了C1和C4的电荷密度,使其α位碳原子的电负性增强,因而酸性催化剂上的H+容易加到噻吩环C1或C4上,生成β位碳正离子(见图1)。
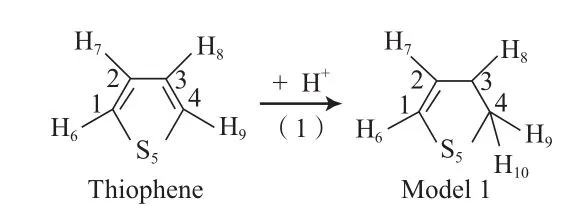
图1 噻吩的质子化Fig.1 The protonation of thiophene.
β位碳正离子在FCC条件下也不稳定,容易转化为其他中间过渡态,再进一步发生C—S键断裂生成H2S,其中涉及噻吩裂化脱硫的中间体主要有:(1)噻吩吸附在催化剂B酸中心,C—S键直接断裂,生成硫醇中间体;(2)噻吩吸附在催化剂B酸中心,通过分子间的氢转移作用使噻吩环饱和,生成硫醚中间体。
Shan等[8]考察了噻吩和烷基噻吩在USY分子筛上的反应过程,认为噻吩裂化脱硫的两个主要步骤是C—S键断裂反应和氢转移反应,并提出了噻吩裂化脱硫机理以及形成中间体的路径(见图2):首先噻吩吸附在B酸中心形成碳正离子,然后发生β键断裂,生成硫醇二烯烃中间体,该碳正离子将继续发生异构化和氢转移反应,使硫醇键进一步断裂生成H2S。Jaimes等[11]也证实了该转化路径的存在。
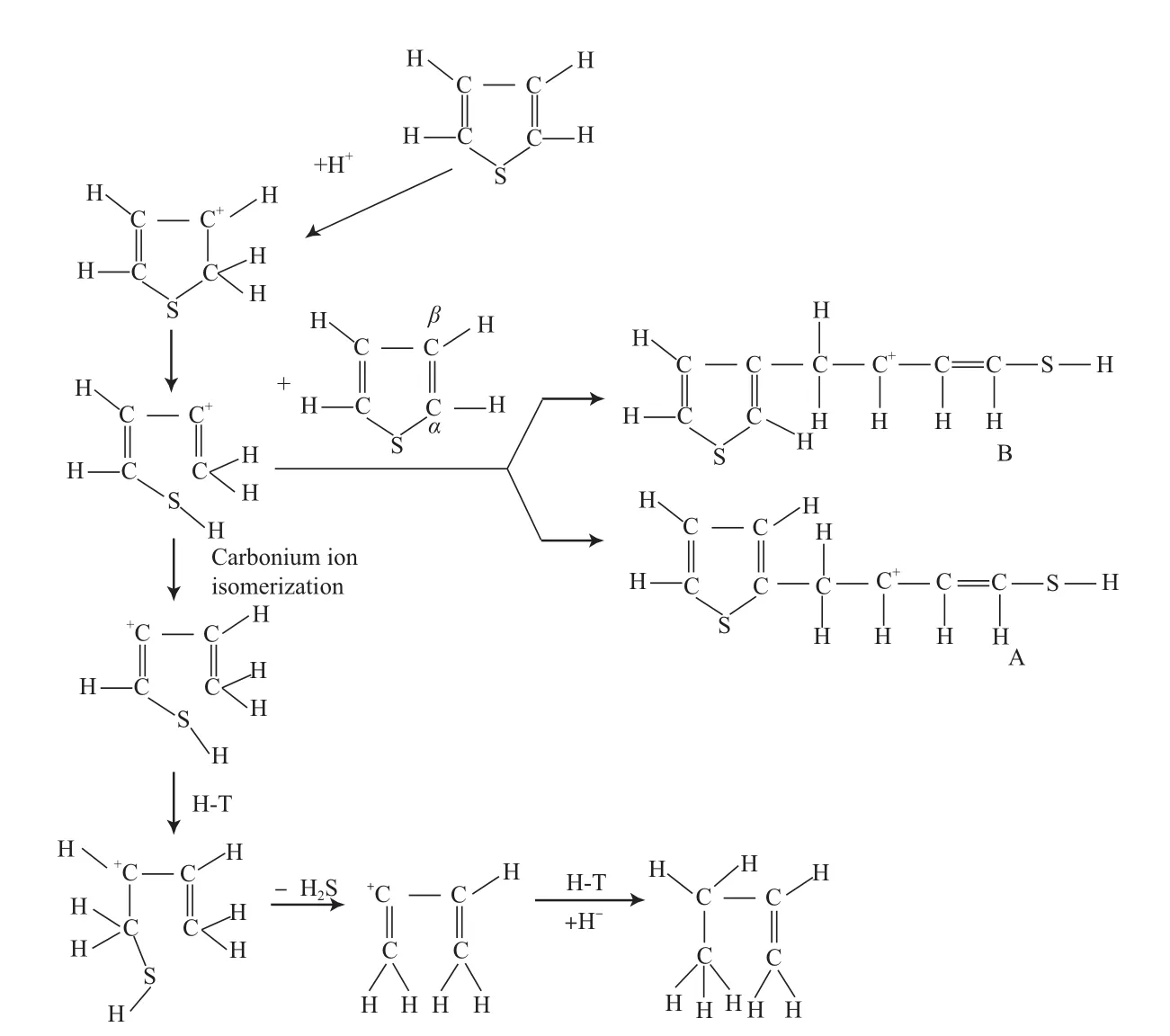
图2 Shan等提出的噻吩裂化脱硫机理[8]Fig.2 Thiophene cracking mechanisms proposed by Shan et al[8].
针对噻吩分子之间的反应,Li等[12]根据密度泛函理论计算,认为噻吩β位碳正离子与另一噻吩分子发生聚合反应后,C—S键才会断裂(见图3),其途径并非如Shan等[8]所述。
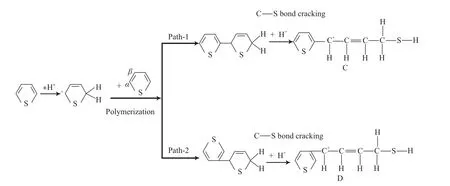
图3 Li等提出的噻吩裂化脱硫机理[12]Fig.3 Thiophene cracking mechanisms proposed by Li et al[12].
此外,噻吩分子之间的聚合反应也存在两条反应路径。经热力学分析表明,噻吩的α位比β位更活跃,故路径1比路径2更具有优势。
对于噻吩在酸性催化剂上通过氢转移作用裂化脱硫的过程,Wang等[13]认为,噻吩在HZSM-5分子筛上反应时首先生成β位碳正离子,再通过氢转移生成具有硫醚性质的物种,当反应温度高于380 ℃时该物种分解为H2S。傅军等[14]和Valla等[15]则认为,当有链烷烃或环烷烃等供氢剂存在时,它们的活泼氢能促进氢转移反应的进行,从而使噻吩环易于饱和,并依次生成二氢噻吩和四氢噻吩,而后者在裂化条件下不稳定,可进一步裂化为丁二烯和H2S。
除裂化脱硫外,噻吩在FCC条件下也很容易缩合生成焦炭。Jaimes等[11]发现,当以辛烷、辛烯、三甲基戊烷和甲基环己烷等为反应溶剂时,噻吩反应产物中焦炭硫的选择性均高达80%以上。此外,在噻吩反应产物中检测到烷基噻吩的存在,并且以辛烯为溶剂时,烷基噻吩的选择性很高,这表明噻吩和烃类之间也发生了烷基化反应。
综上所述,噻吩在FCC过程中的反应路径可能有以下几种:
(1)噻吩的吸附和开环:噻吩吸附在B酸中心,C—S键直接断裂。在没有氢源的情况下,噻吩直接脱硫形成丁二炔和H2S;而在有氢源的情况下,则生成丁二烯和H2S。但由于该路径生成中间物种所需能量较高,因此发生几率很低。
(2)噻吩吸附、开环与另一个噻吩分子聚合:噻吩先吸附开环生成带有双键的硫醇中间体,然后与另一噻吩分子发生聚合反应;经碳正离子的异构化和氢转移,处于β位的C—S键断裂,形成H2S和丁烯基噻吩。
(3)噻吩氢转移和裂化:噻吩经氢转移生成二氢噻吩和四氢噻吩,然后开环断裂生成H2S和丁烯。
(4)噻吩的烷基化:吸附的噻吩与烯烃作用生成烷基噻吩,烷基噻吩继续发生异构化、裂化和环化等二次反应。
(5)噻吩缩合结焦。
此外,苯并噻吩作为FCC原料中一种重要的含硫化合物,它的转化机理和反应路径与噻吩基本相同。朱根权[9]认为苯并噻吩在FCC过程中主要发生以下反应:(1)苯并噻吩的噻吩环直接开环生成H2S和烃类;(2)当反应体系中有供氢剂时,苯并噻吩先加氢饱和生成二氢苯并噻吩再发生开环反应,或与烯烃发生烷基化反应,生成烷基取代的苯并噻吩;(3)苯并噻吩在强酸性中心的催化下,发生缩合结焦反应。与噻吩和苯并噻吩相比,二苯并噻吩在FCC过程的裂化脱硫路径则比较复杂,主要发生缩合结焦反应。
2 不同结构的噻吩类硫化物的反应特点
噻吩可以在纯分子筛上发生反应,但转化率一般很低[16]。甲基环己烷的加入也没有明显提高噻吩的转化率(转化率小于0.2%)[17],故可认为噻吩在FCC条件下一般较稳定。
与噻吩相比,带有侧链的噻吩类硫化物的活性较高,其中侧链的碳数对烷基噻吩的转化率有很大的影响。Corma等[18]也发现噻吩的转化率很低,并且主要生成焦炭;而甲基噻吩由于易形成碳正离子,因此其吸收H-的能力更强,更易于裂化为气体和其他的硫化物进入汽油馏分。Leflaive等[17]的研究结果表明,随侧链碳数的增加,烷基噻吩的转化率明显提高,其中2-甲基噻吩、2-乙基噻吩和2-正己基噻吩的转化率依次为3%,15%,60%;此外,取代基位置对转化率也有影响,与2-正己基噻吩相比,3-正己基噻吩的转化率降至40%左右,这与2位和3位上的电子云密度有关。
侧链的碳数对烷基噻吩反应的选择性也有影响。Corma等[4]认为短侧链(侧链碳数小于等于3)的烷基噻吩主要发生脱烷基和异构化反应,而长侧链的烷基噻吩主要发生侧链裂化和脱氢环化反应;并且侧链碳数越大,侧链环化反应就越易于进行,这与Jaimes等[19]的热力学分析结论相符。但长侧链发生裂化和环化反应的选择性,不仅取决于原料的性质,还与催化剂的性质有关。当催化剂中含有ZSM-5分子筛时,侧链倾向于裂化反应,而环化反应的选择性较低[4]。Leflaive等[17]也研究了侧链的性质对烷基噻吩反应选择性的影响。2-甲基噻吩和2-乙基噻吩均主要发生异构化反应,其中2-乙基噻吩异构化反应的选择性达到55%~65%。但随侧链碳数的增加,异构化反应的选择性逐渐降低,这主要是由于长侧链的烷基噻吩易发生侧链裂化和环化等反应。
此外,侧链数目也是影响烷基噻吩转化的一个重要因素。随侧链数目和侧链上碳数的增加,3-甲基噻吩、2,5-二甲基噻吩和3-丁基噻吩的转化率逐渐提高,在平衡催化剂上的转化率依次为53%,83%,90%,这与碳正离子形成的难易程度有关[20]。但侧链数目对反应类型影响不大,其中3-甲基噻吩和2,5-二甲基噻吩均以异构化反应为主,这与Corma等[4]的结论一致。
Valla等[21]在前期研究[15]的基础上进一步比较了重油中5种典型含硫芳烃的反应性能,转化率高低顺序为:3-癸烷基噻吩>苯并噻吩>二苯并噻吩>4,6-二甲基二苯并噻吩>2-甲基苯并噻吩,其中长侧链的烷基噻吩通过脱烷基和侧链裂化反应,分别生成噻吩和短侧链噻吩而进入汽油馏分,是汽油中硫的主要来源[22];而带有芳环的噻吩对汽油的硫含量影响不大。
3 噻吩类硫化物转化的影响因素
原料类型是决定噻吩类硫化物转化路径和反应活性的决定因素,不同类型分子的裂化产物分布因其分子结构不同而有本质的区别。改变反应体系和反应条件,烃类的转化规律也必然会受到影响,其中反应体系中烃类分子之间的相互作用以及催化剂性质也是影响噻吩类硫化物转化的重要因素。
3.1 烃类的影响
通过分析噻吩的裂化脱硫机理可知,噻吩类硫化物脱硫主要通过氢转移反应进行,Corma等[4]也认为控制噻吩和烷基噻吩裂化反应的关键步骤是氢转移反应。Sara等[23]研究发现,当没有供氢剂时,噻吩的转化率很低,主要发生双分子歧化反应,生成苯并噻吩和H2S;H2的加入也没有明显提高噻吩的转化率,这可能是由于H2在酸性分子筛上不能分解为H+和H-的缘故;与H2相比,丙烷、己烷和癸烷的加入能明显提高噻吩的脱硫速率,其中80%的硫以H2S形式脱除,且噻吩脱硫速率也随供氢剂链长的增加而加快,生成H2S的选择性也逐渐提高,该研究表明大分子的烷烃是很好的供氢剂。
Jaimes等[11]分别以烷烃、烯烃、环烷烃和芳烃为反应溶剂,检测到噻吩的裂化产物中有H2S、烷基噻吩、苯并噻吩和焦炭,这表明烃类分子一方面提供H+,供噻吩开环裂化;另一方面也作为烷基化反应的共同反应物。当以三甲基戊烷、辛烷和甲基环己烷为溶剂时,噻吩裂化产物中H2S的选择性很高,表明直链烷烃和环烷烃是很好的供氢剂,可使噻吩发生开环反应,辛烯则主要与噻吩反应生成烷基噻吩。
与Jaimes等[11]的结论不同,Corma等[4]发现壬烯基本上不参与裂化反应,也不与噻吩发生烷基化反应,但壬烯的加入能促进碳正离子的引发,从而提高噻吩类硫化物的转化速率;而与壬烯的作用正好相反,甲苯的加入明显降低了烷基噻吩和重油的转化率,增加了气体和焦炭的选择性。这主要是因为甲苯在催化剂活性中心上的强吸附作用不利于其他烃类分子和含硫芳烃在活性中心上的吸附和反应。
同一种烃类溶剂对不同噻吩类硫化物的作用也不同。Valla等[15]以十六烷为溶剂,比较了噻吩和苯并噻吩在FCC条件下的反应性能。研究结果表明,噻吩主要进行脱硫反应生成H2S和焦炭,基本不发生烷基化反应;而苯并噻吩的反应活性比噻吩高,主要通过烷基化反应进入沸点更高的馏分中。
3.2 催化剂的影响
催化剂的孔道结构对噻吩类硫化物的反应路径有直接影响。Francisco等[24]发现烷基噻吩和烷基苯并噻吩在稀土Y(REY)和稀土超稳Y(REUSY)分子筛上主要发生侧链裂化反应;而在ZSM-5分子筛上,由于其择形效应影响了噻吩类硫化物的侧链裂化、噻吩环裂化和环化反应的相对反应速率,使长链烷基噻吩裂化为短链烷基噻吩或环化成苯并噻吩的选择性下降,而脱烷基反应的选择性增加,因此一定量ZSM-5分子筛的加入将会增加汽油中的硫含量[25]。
催化剂的酸性和酸类型对噻吩类硫化物的转化和汽油脱硫效果也有影响。朱根全[9]认为催化剂的B酸性强,噻吩和苯并噻吩的转化程度高。这主要是由于强酸性中心有利于它们直接开环,生成H2S和烃类;并且金属中心与酸性中心共同作用也有利于噻吩加氢饱和为二氢噻吩再进一步发生裂化反应。Pang等[26]则认为适量的L酸有利于硫化物的吸附和转化,其中V改性的USY分子筛的脱硫效果最明显。
Robert等[27]研究了噻吩类硫化物在不同酸密度催化剂REY,REUSY,USY/Matrix,USY-G(超稳沸石Z-14G)上的转化规律。研究结果表明,在Y型分子筛上主要存在两种反应机理:(1)催化剂酸密度高,氢转移活性强,则主要发生噻吩的氢转移反应,生成四氢噻吩,并进一步裂化为H2S;(2)催化剂酸密度低,氢转移活性低,则主要发生烷基化反应,生成带有不同烷基侧链的噻吩类化合物,主要有甲基噻吩、乙基噻吩和丙基噻吩等。Lappas等[25]也发现高晶胞参数的催化剂有利于汽油硫含量的降低。当晶胞参数增加0.015 nm,汽油的硫含量降低30%,这主要是由于分子筛的晶胞参数越大,酸密度越大,氢转移作用越强,越有利于噻吩环饱和,从而使其裂化脱硫。
为了降低裂化汽油中的硫含量,众多研究者[24-26,28]考察了脱硫剂或金属对噻吩类硫化物和重油转化的影响。研究结果表明[24,28],GSR-1脱硫剂的加入使烷基噻吩和苯并噻吩侧链的裂化选择性明显降低,而促进了脱氢环化生成苯并噻吩和二苯并噻吩的选择性。这主要由于GSR-1脱硫剂具有较多的L酸中心,对大分子硫化物有很强的吸附能力,有利于环化缩合生成大分子,从而使焦炭和干气的产率增加。金属的加入有利于降低汽油中的硫含量[15,25]。Ni和V易使长侧链的烷基苯并噻吩发生环化反应,并进一步脱氢生成焦炭;而不含金属的催化剂则易于发生烷基化反应,使噻吩和苯并噻吩生成大量的烷基化产物。
4 噻吩类的转化网络
Corma等[4]考察了汽油中不同含硫化合物的转化特点,并建立了汽油馏分中含硫化合物的反应网络。在此反应网络中,H2S是二次不稳定产物,可与反应体系中的其他产物(如烯烃)反应而进一步生成噻吩;而硫醇是一次不稳定产物,很容易裂化生成烃类和H2S。此外,噻吩是一次稳定产物,而烷基噻吩是一次不稳定产物,这两种硫化物不经过氢转移反应则很难发生裂化反应,但由于烷基噻吩易于形成碳正离子中间物种,因而比噻吩的反应速率更快。
根据前人的研究结果和对噻吩类硫化物反应机理的认识,本文建立了典型的噻吩类硫化物的反应网络(见图4)。
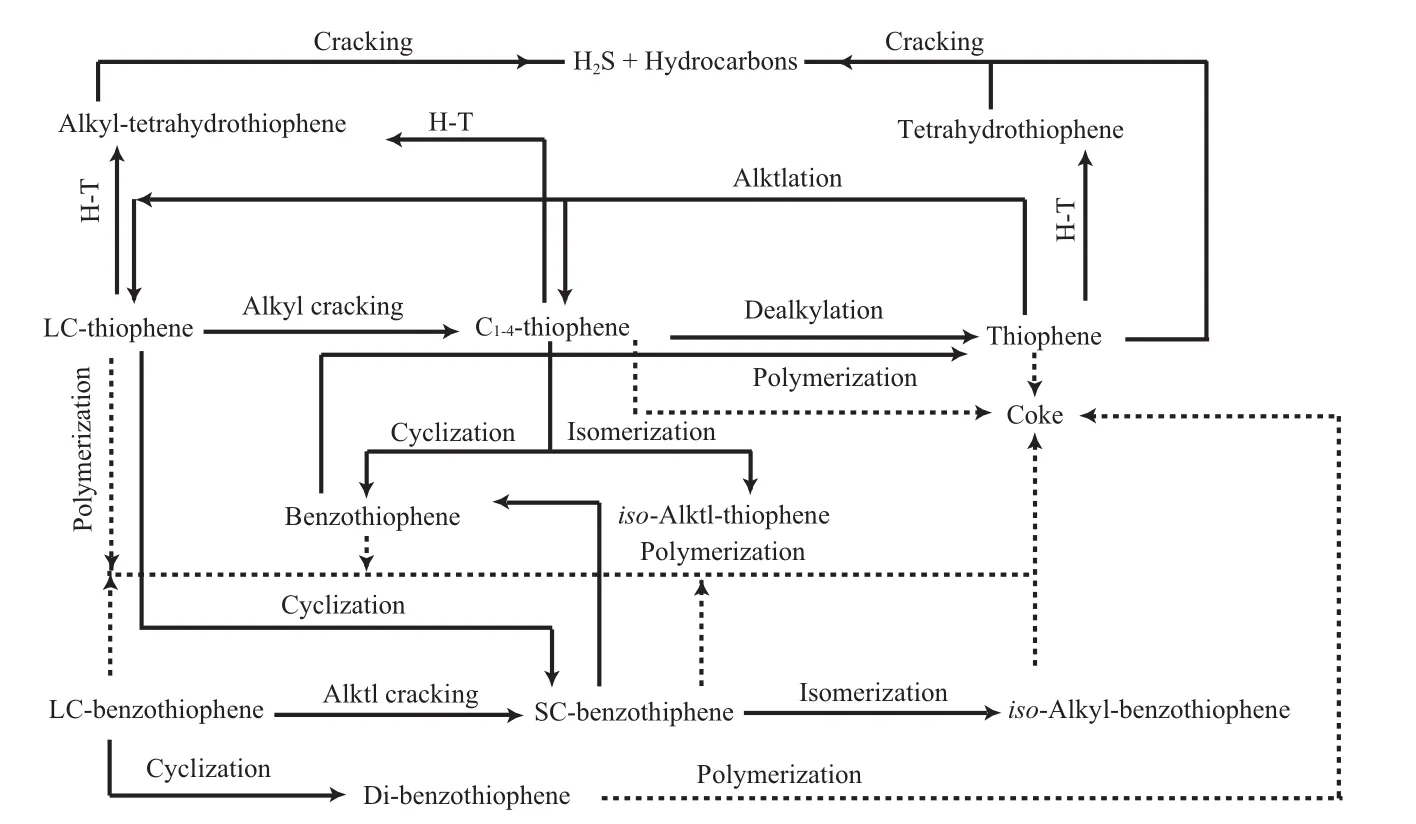
图4 典型的噻吩类硫化物的反应网络Fig.4 The reaction network for conversion of thiophene and thiophene-derivatives.
当反应体系中存在烷烃和烯烃等供氢剂时,噻吩和苯并噻吩在氢转移活性低的催化剂上易于发生烷基化反应,而在氢转移活性高的催化剂上易于发生噻吩环饱和反应,并进一步裂化脱硫;而对于带烷基侧链的噻吩类硫化物,由于易生成碳正离子,它们的活性较高,其中短侧链的烷基噻吩和苯并噻吩易发生异构化反应和脱烷基反应,而长侧链的烷基噻吩和烷基苯并噻吩则易于发生侧链裂化和环化反应。当催化剂中含有金属或脱硫剂时,不管烷基侧链存在与否,噻吩和苯并噻吩类硫化物都很容易发生聚合生焦反应。
5 结语
研究者对于典型的噻吩分子在FCC过程中的转化机理已有初步认识,但由于分离和分析技术的限制,对实际石油体系中含硫芳烃的分子结构缺乏全面认识,导致对含硫芳烃的转化规律认识不深入。随着资源、市场和环保等方面的压力和要求的不断提高,在分子水平上研究原料油的烃类结构组成及其转化规律已成为当前FCC技术发展的方向和必然要求。因此,未来的研究工作还需加强对含硫芳烃结构的分析,同时在分子水平上深入研究不同类型的含硫芳烃的转化规律,以及考察原料中含硫化合物的组成分布与产物分布的关系。只有充分掌握原料中的含硫化合物在FCC过程中的转化规律,才能有效控制含硫化合物的转化,生产更加清洁的燃料。
[1] 汤海涛,凌珑,王龙延. 含硫原油加工过程的硫转化规律[J]. 炼油设计,1999,29(8):9 - 15.
[2] 殷长龙,夏道宏. 催化裂化汽油中类型硫含量分布[J]. 燃料化学学报,2001,29(3):256 - 259.
[3] 王征,杨永坛. 柴油中含硫化合物类型分布及变化规律[J].分析仪器,2010(1):70 - 73.
[4] Corma A,Martinez C,Ketley G,et al. On the Mechanism of Sulfur Removal During Catalytic Cracking[J].Appl Catal,A,2001,208(1/2):135 - 152.
[5] Dupain X,Rogier E,Jacob A,et al. Cracking Behavior of Organic Sulfur Compounds Under Realistic FCC Conditions in a Microriser Reactor[J].Appl Catal,A,2003,238(2):223 - 238.
[6] 袁海亮. 噻吩硫裂化规律及改性分子筛脱硫性能的研究[D].北京:石油化工科学研究院,2001.
[7] 郑柯文,高金森,徐春明. 噻吩催化裂化脱硫机理的量子化学分析[J]. 化工学报,2004,55(1):87 - 90.
[8] Shan Honghong,Li Chunyi,Yang Chaohe,et al. Mechanistic Studies on Thiophene Species Cracking over USY Zeolite[J].Catal Today,2002,77(1/2):117 - 126.
[9] 朱根全. 催化裂化条件下含硫化合物转化规律的研究[D]. 东营:中国石油大学(华东),2001.
[10] Kissin Y V. Chemical Mechanisms of Catalytic Cracking over Solid Acidic Catalysts: Alkanes and Alkenes[J].Catal Rev,2001,43(1/2):85 - 146.
[11] Jaimes L,Badillo M,Lasa H. FCC Gasoline Desulfurization Using a ZSM-5 Catalyst Interactive Effects of Sulfur Containing Species and Gasoline Components[J].Fuel,2011,90(5):2016 - 2025.
[12] Li Bingrui,Guo Wenping,Yuan Shuping,et al. A Theoretical Investigation into the Thiophene-Cracking Mechanism over Pure Brönsted Acidic Zeolites[J].J Catal,2008,253(1):212 - 220.
[13] Wang Xiangsheng,Luo Guohua. The Removal of Thiophene from Coking Benzene over HZSM-5 Zeolite[J].Chin J Catal,1996,17(6):530 - 534.
[14] 傅军,王鹏,何鸣元. 含噻吩烃类在沸石上的裂化脱硫[J].石油学报:石油加工,2002,18(3):36 - 42.
[15] Valla J A,Lappas A A,Vasalos I A. Catalytic Cracking of Thiophene and Benzothiophene Mechanism and Kinetics[J].Appl Catal,A,2006,297(1):90 - 101.
[16] Welters W J J,Beer V H J,Santen R A. Influence of Zeolite Acidity on Thiophene Hydrodesulfurization Activity[J].Appl Catal,A,1994,119(2):253 - 269.
[17] Leflaive P,Lemberton J U,Perot G,et al. Formation of Sulfur-Containing Compounds Under Fluid Catalytic Cracking Reaction Conditions[J].Stud Surf Sci Catal,2000,130:2465 - 2470.
[18] Corma A,Gullbrand P,Martinez C. Gasoline Sulfur Removal Kinetics of S Compounds in FCC Conditions[J].Stud Surf Sci Catal,2001,134(2):153 - 165.
[19] Jaimes L,Lujan M,Lasa H. Thiophene Conversion Under Mild Conditions over a ZSM-5 Catalyst[J].Chem Eng Sci,2009,64(11):2539 - 2561.
[20] Valla J A,Lappas A A,Vasalos I A. On the Mechanism and Kinetics of Alkylthiophenes During the FCC Process[R]//Workshop of CPERI 2004,Thessaloniki:CPERI,2004:60 - 70.
[21] Valla J A,Mouriki E,Lappas A A,et al. The Effect of Heavy Aromatic Sulfur Compounds on Sulfur in Cracked Naphtha[J].Catal Today,2007,127(1/4):92 - 98.
[22] Leflaive P,Lemberton J L,Pérot G,et al. On the Origin of Sulfur Impurities in Fluid Catalytic Cracking Gasoline—Reactivity of Thiophene Derivatives and of Their Possible Precursors Under FCC Conditions[J].Appl Catal,A,2002,227(1/2):201 - 215.
[23] Sara Y,Toshio W,Enrique I. Catalytic Desulfurization of Thiophene on H-ZSM5 Using Alkanes as Co-Reactants[J].Appl Catal,A,2003,242(1):111 - 121.
[24] Francisco H B,Juan C M M,Roberto Q S,et al. Sulfur Reduction in Cracked Naphtha by a Commercial Additive:Effect of Feed and Catalyst Properties[J].Appl Catal,B,2001,34(2):137 - 148.
[25] Lappas A A,Valla J A,Vasalos I A,et al. The Effect of Catalyst Properties on the in Situ Reduction of Sulfur in FCC Gasoline[J].Appl Catal,A,2004,262(1):31 - 41.
[26] Pang Xinmei,Zhang Li,Sun Shuhong,et al. Effects of Metal Modifications of Y Zeolites on Sulfur Reduction Performance in Fluid Catalytic Cracking Process[J].Catal To-day,2007,125(3/4):173 - 177.
[27] Robert R G,Robert H H,Thomas G A,et al. Influence of Catalyst on Sulfur Distribution in FCC Gasoline[J].Div Fuel Chem,1992,37(1):33 - 40.
[28] Francisco H B,Roberto Q S,Jaime S V. Effect of Highly Reactive Sulfur Species on Sulfur Reduction in Cracking Gasoline[J].Appl Catal,B,2003,42(2):145 - 154.
Research Progresses in Conversion of Thiophene Derivatives in FCC Process
Wu Qunying,Da Zhijian,Zhu Yuxia
(SINOPEC Research Institute of Petroleum Processing,Beijing 100083,China)
Cracking desulfurization mechanisms of thiophene and its derivatives in FCC process were reviewed. The effect of molecular structures of thiophene and its derivatives on the cracking desulfurization was discussed in detail. Many researches showed that the thiophene conversion was slow under the FCC conditions. The reaction activities of thiophene derivatives with side chains were higher than that of thiophene. Alkyl-thiophene derivatives with short side chain trend to isomeration and dealkylation,and alkyl-thiophene derivatives with long side chain trend to cracking and cyclisation of the side chains. The effects of the properties of both the hydrocarbons and catalysts on the cracking desulfurization of thiophene derivatives were evident. The hydrogen donor,such as alkanes and cycloparaffins,and the catalysts with high hydrogen transfer activity were conducive to the cracking desulfurization. Based on the above discussion,a reaction network for the cracking desulfurization of typical thiophene derivatives was proposed.
thiophene;benzothiophene;fluid catalytic cracking;hydrogen transfer
1000-8144(2012)04 - 0477 - 07
TE 624.4
A
2011 - 09 - 21;[修改稿日期]2011 - 12 - 20。
吴群英(1983—),女,湖北省黄石市人,博士生,工程师,电话 010-82368917,电邮 qunying2005@126.com。
(编辑 王 萍)
