光反应合成异吲哚酮并环苄基甘氨酸肽*
2012-09-17庄丽梅侯云雷王丽杰金英学
庄丽梅,姜 鹤 ,侯云雷,王丽杰,金英学
(哈尔滨师范大学)
0 引言
环肽化合物由于两个端基得到保护,增强了抗酶降解能力,提高了生物利用度.同时,因为环肽构象限定,能更好地与受体结合,所以增强了生物活性.环肽广泛分布在植物、海洋生物、微生物和少部分动物体中,但含量极少,而且分离困难.因此环肽的合成成为肽化学领域当前要解决的首要问题.文献报道用光诱导单电子转移(Single Electron Transfer简称 SET)环化反应合成冠醚方法[1-4],笔者用此方法合成了异吲哚酮并环苄基甘氨酸二肽,一直到异吲哚酮并环苄基甘氨酸五肽[5-9].该文在前期工作基础上合成了异吲哚酮并环苄基甘氨酸六肽到环七肽.同时合成了氨基酸氮上有自由氢的环甘氨酸二肽和三肽.合成路线如图1所示.以相应的氨基酸为起始原料,经过氨基酸缩合反应得到氨基酸肽链(1a,1b),再与N-邻苯二甲酰甘氨酰氯缩合得到N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺(2a,2b),在甲醇中进行光反应最终得到异吲哚酮并环苄基甘氨酸肽(3a,3b).
1 实验部分
Varian-400型核磁共振仪(TMS为内标)(美国 瓦里安公司);Hitachi VG-7070型质谱仪(日本日立公司);450 W中压汞灯做反应光源(德国贺利氏公司);2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉(简称 EEDQ)、N-t-Boc-苄基甘氨酸、苄基甘氨酸均购自Aldrich公司,其它试剂由国药集团提供;所有反应均在氮气保护下进行;所有有机溶剂均经干燥纯化.
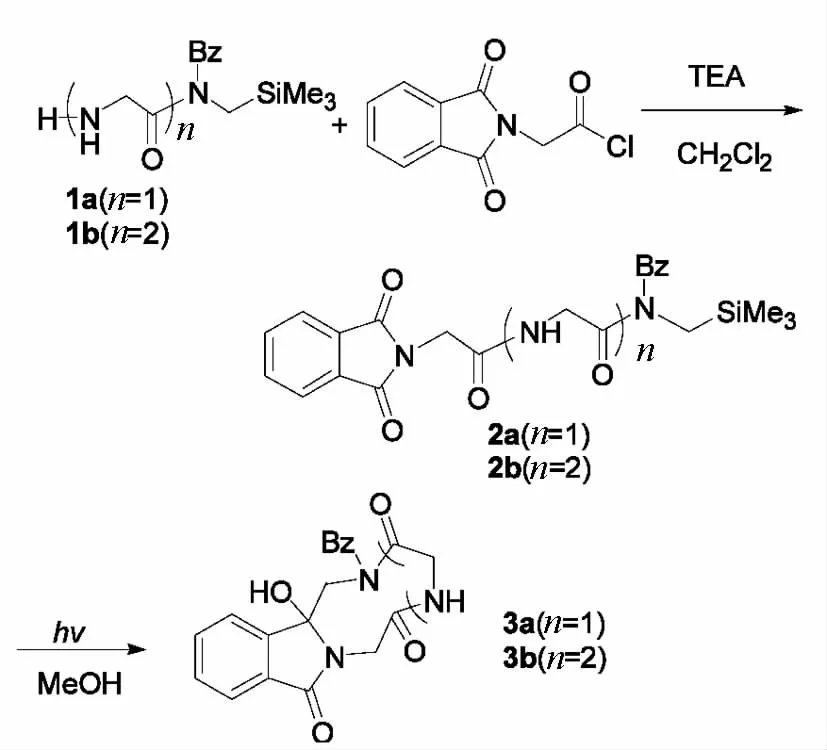
图1 光反应物3的合成
1.2 N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺(2a,2b)的合成
将末端三甲基硅甘氨酸肽链1(7.5 mmol)溶于100 mL干燥的二氯甲烷中,分别加入三乙胺(3.9 mL,22.5 mmol),边搅拌边分别加入 N-邻苯二甲酰甘氨酰氯(1.68 g,7.5 mmol),室温搅拌3 h,减压蒸馏除去二氯甲烷,残余物分别用硅胶柱层析(洗脱剂:V乙酸乙酯/V正己烷=2∶1),分离得纯的产物2a,2b.
2a 产率 80%.1H NMR(CDCl3)δ:0.03(s,9H,SiMe3),2.91(s,2H,CH2SiMe3),4.10(s,2H,CH2CON(CH2Ph)CH2SiMe3),4.40(s,2H,CON(CO)CH2),4.43(S,2H,CH2Ph);13C NMR(CDCl3)δ:-1.2,38.6,40.2,41.1,52.0,123.2,126.1,127.3,127.4,128.9,131.8,134.0,135.1,165.8,166.6,167.6;FAB-MSm/z437.26(M+).
2b 产率80%.1H NMR(CDCl3)δ:0.02(s,9H,SiMe3),2.89(s,2H,CH2SiMe3),4.02(s,2H,CH2CON(CH2Ph)CH2SiMe3),4.23(s,2H,CH2CONHCH2CON(CH2Ph)CH2SiMe3),4.32(s,2H,CON(CO)CH2),4.44(S,2H,CH2Ph);13C NMR(CDCl3)δ:-1.2,38.4,39.9,41.1,41.3,52.1,123.2,126.3,127.2,127.4,128.6,132.3,134.0,135.2,165.6,166.5,166.7,167.6;MS(FAB)m/z494.18(M+)
1.3 异吲哚酮并环肽(3a,3b)的合成
将化合物3(0.5 mmol)溶解在150 mL甲醇中,置于光反应器内,通入氮气流30 min,彻底驱除反应体系内的空气,在氮气保护下分别用波长290 nm紫外光照射1 h,反应物100% 转化.撤掉光源,浓缩反应液,残余物用50 mL二氯甲烷稀释,加入30 mL 15% 的氯化钠溶液分层,有机层用无水硫酸钠干燥、浓缩,残余物分别用硅胶柱层析分离提纯(洗脱剂:V乙酸乙酯/V正己烷=2∶1)得目标产物 3a,3b.
3a无色油状物,产率45%;1H NMR(CDCl3)δ:3.27(d,J=12.4 Hz,1H,C(OH)CH),4.01(d,J=12.4 Hz,1H,C(OH)CH),4.55(s,2H,NCH2CO),4.65(s,2H,CH2Ph),7.12 ~7.78(m,9H,Ar);13C NMR(CDCl3)δ:41.0,51.1,54.1,83.5,123.1,126.1,127.4,127.5,128.1,128.2,129.6,131.4,134.5,135.8,162.5,163.1;FAB-MSm/z366.1(M++1).
3b无色油状物,产率 38%;1HNMR(CDCl3)δ:3.43(d,J=12.7 Hz,1H,C(OH)CH),4.11(d ,J=12.7 Hz,1H,C(OH)CH),4.35(s,2H,NCH2CONHCH2),4.38(s,2H,NCH2CO),4.59(s,2H,CH2Ph),7.12 ~7.78(m,9H,Ar);13C NMR(CDCl3)δ:41.0,51.1,51.3,53.6,83.6,123.3,126.0,127.4,127.6,127.7,128.2,130.2,131.1,135.0,135.8,162.3,162.5,163.1;MS(FAB)m/z422.2(M++1).
2 结果与讨论
2.1 N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺(2a,2b)的合成
以市售的氨基酸为起始原料,按文献[7]方法制备N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺(2a,2b).一般方法如下:将末端三甲基硅甘氨酸溶于适量的干燥的四氢呋喃中,加入等摩尔的EEDQ,加入等摩尔的Boc-甘氨酸,回流12 h,冷却至室温,减压蒸馏除去四氢呋喃,加入适量的水,用适量的乙酸乙酯萃取,合并、浓缩有机层,残余物溶于适量的二氯甲烷中,滴入适量的三氟乙酸,室温搅拌2 h,用饱和碳酸钠溶液中和至中性,用适量乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,有机层用无水硫酸钠干燥,浓缩得到末端三甲基硅甘氨酸肽链1a.类似地重复上述过程得到目标甘氨酸肽链1b.末端三甲基硅甘氨酸肽链(1a,1b)在干燥的二氯甲烷中,三乙胺存在下与N-邻苯二甲酰甘氨酰氯缩合,室温搅拌3 h,减压蒸馏除去二氯甲烷,残余物用硅胶柱层析(洗脱剂:V乙酸乙酯/V正己烷=2∶1)分离得纯的产物2a,2b.
2.2 光诱导单电子转移反应机理
N-(末端三甲基硅苄基甘氨酸肽链)邻苯二甲酰亚胺是分子内给受电子体系,邻苯二甲酰亚胺基为电子受体(A),羰基为受体的发色基团,甘氨酸肽链为电子给体(D).电子受体A光致激发,激发态A*与基态D构成激基缔合物[A*-D],A*的HOMO轨道上的一个电子跃迁到LUMO轨道,HOMO轨道变成 SOMO,LUMO轨道变成SOMO'.基态D的HOMO轨道上一个电子转移到激发态A*的SOMO轨道上,形成分子内双离子自由基对[A-·-D+·].图 2中ⅠPD为电子给体D的电离势,EAA*为电子受体A激发态A*的亲和势,E(D·+/D)为基态D的氧化电位,E(A·-/A*)为激发态A*的还原电位.激发态A*与基态D发生自发的电子转移必须使自由能变化 ΔG=ⅠPD-EAA*< 0,因此 ΔG=E(D·+/D)-E(A·-/A*)< 0.酰胺氮原子为电子给体时,氧化电位E(D·+/D)=V,邻苯二甲酰亚胺激发态为电子受体时,还原电位E(A·-/A*)=1.6V,E(D·+/D)-E(A·-/A*)<0,N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺能够在光诱导下能够自发地发生电子转移的氧化还原反应,生成分子内双离子自由基,自由基发生化学反应(CR).
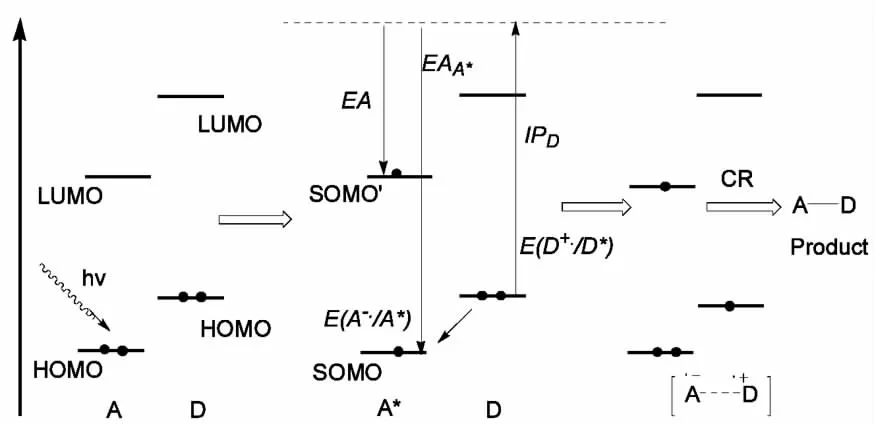
图2 A*与D的氧化还原反应中与电子转移有关能级及电子转移示意
2.3 N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺的光反应
如图3所示,由邻苯二甲酰亚胺和甘氨酸肽链构成的分子内给受电子体系N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺(1~3)受光照射,受体发色团光致激发态与相邻的基态给电子杂原子氮发生首次单电子电子转移(SET),生成双离子自由基(4),电荷在肽链上沿着化学键传递(ISET),形成共振杂化体[4-6],链上末端离去基团-SiMe+3离去,形成分子内端位双自由基7,双端位自由基7分子内迅速偶合生成带-OS-iMe3机的异吲哚酮并环苄基甘氨酸肽(8).在甲醇溶剂(质子性溶剂)中SiMe3基团与质子置换形成带羟基的异吲哚酮并环苄基甘氨酸肽(3).

图3 光反应历程
3 结束语
以甘氨酸为起始原料,合成了2个新的光反应物,即N-(末端三甲基硅甘氨酸肽链)邻苯二甲酰亚胺,在甲醇溶剂中进行了光反应得到了2个新型的异吲哚酮并环苄基甘氨酸肽.虽然产率一般,却给我们指出了一种合成特殊结构环肽化合物的方法.
[1] Kanaoka,Y,Koyama,K,Flippen J L,et al.Photochemistry of the phthalimide systemⅥ:Photocyclization of N-alicyclic phthalimides.Synthesis of multicyclic benzazepine systems[J]J Am Chem Soc,1974,96(4):4719-4721..
[2] Yoon U C,Mariano P S.Mechanistic and Synthetic Aspects of Amine-Enone Single Electron-Trasfer Photochemistry[J].Acc Chem Res ,1992,25(3):233-240.
[3] Cho D W,Choi J H,Oh S W,et al.Single Electron Transfer-Promoted Photocyclization Reactions of Linked Acceptor Polydonor Systems:Effects of Chain Length and Type on the Efficiencies of Macrocyclic Ring-Forming Photoreactions of Tethered?-Silyl Ether Phthalimide Substrates.J Am Chem Soc,2008,130(7):2276-2284.
[4] Sung N K.Cho D W.Choi J H,et al.Mariano P S A Facile Approach to the Preparation of Bis-Crown Ethers Based on SET-Promoted Photomacrocyclization Reactions.J Org Chem,2007,72(23):8831-8837.
[5] Yoon U C,Jin Y X,Oh S H,et al.Comparison of Photomacrocyclization Reactions of Trimethylsilyl-and Tributylstannyl Terminated Phthalimido–and Maleimido-polyethers.Journal of Photochemistry and Photobiology A:Chemistry[J],2002.150(1),77-84.
[6] Yoon U C,Jin Y X,Oh S W,et al.A Synthetic Strategy for the Preparation of Cyclic Peptide Mimetics Based on SETPromoted Photocyclization Processes[J].J Am Chem Soc,2003,125(12):10664-10671.
[7] Jin Yi X,Tan G H,Liu S S.Photoinduced Cyclization Reactions of N-(ω-teributyltinalkyl)phthalimides[J].Chin J Synth Chem,2008,16(2):153-157.
[8]Jin Y X,Tan G H,Wang T C,et al.Photoinduced Cyclization Reactions of Phthalimide Derivatives N-Linked Two Electrondonating Chains by Single Electron Transfer[J].Chin J Org Chem,2010,30(4):595-600.
[9] Jin Y X,Yuan W,Qu F Y,et al.Photoinduced Single Electron Transfer Cyclization Reactions of N-(ω-tributylstannylpolyethoxyether)phthalimides[J].Acta Chim Sinica,2011,69(20):2407-2412.
