集胞藻PCC6803膜脂循环关键基因酶学性质和生理功能
2012-09-03高倩倩谈晓明吕雪峰
高倩倩,谈晓明,吕雪峰,3
1 中国科学院青岛生物能源与过程研究所 中国科学院生物燃料重点实验室,山东 青岛 2661012 中国科学院研究生院,北京 1000493 山东省能源生物遗传资源重点实验室,山东 青岛 266101
集胞藻PCC6803膜脂循环关键基因酶学性质和生理功能
高倩倩1,2,谈晓明1,吕雪峰1,3
1 中国科学院青岛生物能源与过程研究所 中国科学院生物燃料重点实验室,山东 青岛 266101
2 中国科学院研究生院,北京 100049
3 山东省能源生物遗传资源重点实验室,山东 青岛 266101
高倩倩, 谈晓明, 吕雪峰. 集胞藻 PCC6803膜脂循环关键基因酶学性质和生理功能. 生物工程学报, 2012, 28(12):1473−1481.
Gao QQ, Tan XM, Lü XF. Characterization of a key gene in membrane lipid cycle inSynechocystissp. PCC6803. Chin J Biotech, 2012, 28(12): 1473−1481.
集胞藻PCC6803野生型和其脂酰ACP合酶敲除突变株的自由脂肪酸含量和组成表明膜脂的重构和降解是细胞内自由脂肪酸的来源之一。在这一过程中脂肪酶起到关键性作用。通过基因组数据库检索,发现集胞藻PCC6803基因组中只有一个脂肪酶编码基因sll1969,但是还没有其功能相关的生化证据。为了确定该基因的功能及其在脂肪酸代谢途径中的作用,加深对集胞藻PCC6803脂肪酸代谢途径的了解,文中将sll1969基因在大肠杆菌中过表达和体外纯化,得到重组蛋白Sll1969,并对其酶学性质进行初步分析。在30 ℃条件下,测得Sll1969以对硝基苯丁酸酯作为底物时的Km和kcat值分别为 (1.16±0.01) mmol/L和 (332.8±10.0)/min;该脂肪酶的最适反应温度为55 ℃。通过比较分析sll1969突变株中脂肪酸含量和组成变化,发现sll1969的表达量与细胞自由脂肪酸的产量呈正相关,但Sll1969不是细胞中唯一的脂肪酶。
脂肪酶,sll1969,对硝基苯丁酸酯,脂肪酸
面对日益增长的能源需求和日益严峻的温室效应问题,可再生的环境友好型的生物能源正逐渐凸显其巨大的应用潜力。脂肪酸族生物燃料因其能量密度高,与现行的运输系统兼容性好等优势而逐渐受到人们的关注,关于脂肪酸族生物燃料在基因工程大肠杆菌的生物合成也成为研究热点[1-3]。放氧光合微生物蓝细菌由于具有生长速度快、遗传操作简单等优势,近年来也被用于包括脂肪酸族生物燃料在内的多种生物燃料和生物化学品分子的生物合成研究[4-5]。
Schirmer等首次鉴定了蓝细菌中的脂肪烃生物合成途径:脂酰 ACP被脂酰 ACP还原酶(AAR) 还原为脂肪醛,脂肪醛被脂肪醛脱羰基酶(ADC) 催化最终形成脂肪烃[6]。谈晓明等通过在集胞藻PCC6803中表达不同来源的脂酰CoA还原酶基因 (far) 实现了脂肪醇在蓝细菌的生物合成,最高产量达到200 μg/L[7]。
蓝细菌生物合成脂肪醇和脂肪烃等脂肪族生物液体燃料包括两个关键步骤:过量生产脂肪酸和脂肪酸衍生化[4]。为了在蓝细菌中大量生产脂肪酸,刘欣尧等通过过表达集胞藻 PCC6803本身的脂酰 CoA羧化酶和外源的硫脂酶,以及敲除碳源竞争途径 PHB合成等途径和细胞S-layer合成途径中相关基因等基因工程改造工作,最终实现了自由脂肪酸的过量生产和分泌,最高产量达到 (197±14) mg/L[8]。随后,刘欣尧等又通过条件诱导启动子控制外源脂肪酶的表达,使之在产脂肪酸细胞培养平台期降解细胞膜脂,从而进一步释放和提高自由脂肪酸的产量[9]。最近,美国桑迪亚国家实验室的一个研究小组也实现了在另一种蓝细菌聚球藻 PCC7942中合成和分泌自由脂肪酸[10]。这一系列工作表明基因工程蓝细菌脂肪族类液体生物燃料产量的提高还有很大的空间。
关于集胞藻PCC6803脂酰ACP合酶 (aas)突变株的同位素标记示踪实验表明,集胞藻体内存在一个特殊的自由脂肪酸到膜脂的循环途径:即细胞膜脂能够被未知的脂肪酶水解,释放出自由脂肪酸;自由脂肪酸被脂酰 ACP合酶重新激活为脂酰ACP,后者又被酰基转移酶重新利用而掺入到细胞膜脂上[11-12]。在这一过程中,将细胞膜脂水解为自由脂肪酸的脂肪酶发挥了重要的生理功能。而鉴定脂肪酶编码基因和解析该脂肪酶的生理功能,对进一步解析蓝细菌膜脂循环途径的生理功能以及进一步提高基因工程蓝细菌脂肪族生物燃料的产量具有重要的意义。
针对集胞藻 PCC6803基因组中唯一的脂肪酶候选基因sll1969,本研究开展了一系列有关的体外酶学表征以及生理功能鉴定工作,最终确定该基因编码蛋白为脂肪酶,但是它不是集胞藻PCC6803基因组中唯一的脂肪酶基因。
1 材料与方法
1.1 试剂
十九烷酸购自Sigma-Aldrich公司 (美国)。其他化学试剂购自Merck公司 (德国) 或Amresco公司 (美国)。TaqDNA聚合酶和限制性内切酶购自Fermentas公司 (加拿大) 或者TaKaRa公司(日本)。用于分子克隆的试剂盒购自 Omega或TaKaRa (日本)。DNA引物在Sangon (中国上海) 合成。DNA分子标记购自TaKaRa公司(日本)。
1.2 质粒和突变菌株的构建
以集胞藻PCC6803基因组DNA为模板,以Sll1969ndes/Sll1969xhoIcas和 Sll1969bglIIs/Sll1969xhochas (表1) 为引物对,PCR得到两端带有不同酶切位点的DNA片段;按照分子克隆实验手册[13]的方法,分别将这两个片段插入表达载体pET21b (Novagen, 美国)NdeⅠ/XhoⅠ位点和pXT37b[7]的BglⅡ/XhoⅠ位点,分别得到质粒pGQ73和pGQ48。
以集胞藻PCC6803基因组DNA为模板,分别利用引物对 1969kuF/R和 1969kdF/R扩增sll1969上下游各500 bp左右的片段,并分别将它们克隆到pMD18-T载体,得到质粒pGQ43和pGQ44。以BamHⅠ将质粒 pRL446[14]携带的的卡那霉素抗性基因片段C.K2切下,插入经过同样酶切的pGQ43,得到质粒pGQ45。之后,pGQ45经过DraⅠ和EcoRⅠ酶切补平后,回收1 700 bp左右的片段,插入经过SmaⅠ酶切的pGQ44,得到质粒pGQ46。所有载体构建经过酶切或者测序验证。
鉴定正确的质粒pGQ48和pGQ46,参照文献报道的转化方法[15],转化集胞藻PCC6803aas敲除突变株 GQ8[16]后,分别得到菌株 GQ12和GQ13。GQ12转化子用携带5 mg/L壮观霉素和5 mg/L红霉素的抗性平板筛选得到;GQ13转化子用携带5 mg/L卡那霉素和5 mg/L红霉素的BG11平板筛选得到。整合完全的突变株通过基因组PCR加以验证。
1.3 蛋白表达和纯化
将构建得到的蛋白表达质粒 pGQ73导入到大肠杆菌BL21 (DE3) 中。挑取转化子,经过液体过夜培养后,以 1∶100的比例稀释到新的液体LB培养基中,37 ℃培养。待OD600达到0.6~0.7时,加入0.2 mmol/L IPTG,之后转到16 ℃进行蛋白诱导表达 12 h。离心收集细胞 (5 000×g,4 ℃,10 min),收集的细胞重悬于结合缓冲液(20 mmol/L PBS,0.5 mol/L NaCl,pH 6.9),冰浴中超声破碎。破碎完全后,离心 (14 000×g,4 ℃,30 min) 并收集上清。上清液经过0.22 μm滤膜过滤后上镍柱纯化,纯化过程按照蛋白纯化说明书 (Novagen) 执行。纯化得到的蛋白经过SDS-PAGE和 Western blotting验证。Western blotting检测采用PVDF膜 (Roch,美国)。利用6×His-标签探针标记目的蛋白,最终使用碱性磷酸酶显色试剂盒 (Amresco,美国) 进行检测。蛋白浓度的测定方法参照Bradford方法[17]。

表1 本研究中所用的引物Table 1 Primers used in this study
1.4 脂肪酶活性检测
脂肪酶活性检测参照文献报道的方法[18]进行,反应体系使用对硝基苯丁酸酯作为底物。脂肪酶催化底物分解产生对硝基苯,在此过程中不断测定光吸收值的变化。根据单位时间产生对硝基苯酚的量来反应酶活力。反应A液:3 g/L的对硝基丁酸酯的异丙醇溶液 (冷藏),B液含有0.4% TritonX-100和 0.1%阿拉伯树胶的0.1 mol/L PBS缓冲液 (pH 7.0)。测定时将A、B两种储存液按照1∶9的体积比混合。取900 μL混合液加入100 μL适当浓度的酶液混匀测定吸光值 (410 nm) 的变化。在此反应条件下对硝基苯的消光系数是 1.5×104L/(mol·cm)。
1.5 菌株的培养和自由脂肪酸的提取测定
集胞藻菌株在 30 ℃条件下进行光照培养(30 μE /(m2·s)),突变株 GQ8 在含有 10 mg/L 红霉素的BG11培养基[19]中培养;突变株GQ12在含有10 mg/L壮观霉素和10 mg/L红霉素的BG11中培养;突变株GQ13在含有10 mg/L卡那霉素和10 mg/L红霉素的BG11中培养。用于自由脂肪酸测定的菌株于50 mL液体BG11中,摇床培养。收集生长至平台期的或平台期后转至42 ℃热激1 d后的藻细胞进行自由脂肪酸提取和测定[20]。
2 结果与分析
2.1 基因的异源表达、蛋白纯化及部分酶学性质分析
经过测序和酶切鉴定正确的sll1969基因表达载体 pGQ73 (图 1),被导入大肠杆菌 BL21(DE3) 进行异源表达。37 ℃,0.2 mmol/L IPTG诱导3 h时,得到的目的蛋白主要以包涵体的形式存在。而采用16 ℃诱导时,得到的可溶性蛋白含量增加。最终选择以16 ℃诱导过夜的细胞用于蛋白纯化。sll1969基因编码蛋白预测分子量为22 kDa,纯化的蛋白携带His标签预测总大小为 23.5 kDa。纯化得到的蛋白经过 12%浓度的SDS-PAGE和Western blotting检测 (图2),与预测的蛋白大小基本相符。纯化得到的蛋白经过脱盐和30% PEG20000浓缩后,利用Bradford方法进行蛋白定量。最终,在1 L的培养物中共纯化得到约2.5 mg蛋白。以对硝基苯丁酸酯作为底物进行酶学性质分析,最终测得30 ℃条件下该酶Km和kcat值分别为(1.16±0.01) mmol/L 和332.8±10.0/min。酶活性的温度的依赖性检测表明酶的最适反应温度为55 ℃ (图2)。
2.2 sll1969突变株的构建
为了研究脂肪酶 (Sll1969) 在集胞藻PCC6803脂肪酸代谢途径中的作用,作者分别构建了敲除sll1969基因和过表达sll1969基因的两种突变株。首先构建了用于sll1969基因敲除的质粒 pGQ46和用于sll1969基因过表达的质粒pGQ48 (图 1)。通过同源重组[15],质粒 pGQ46携带的卡那霉素抗性基因能够替换基因组中的sll1969基因编码区;而质粒pGQ48携带的PpetE启动子[21]驱动的sll1969基因表达元件将插入到集胞藻PCC6803中性位点slr0168[15]位点,实现该基因过表达。将质粒pGQ46和pGQ48分别转化aas敲除突变株GQ8[16]得到菌株GQ13和GQ12。
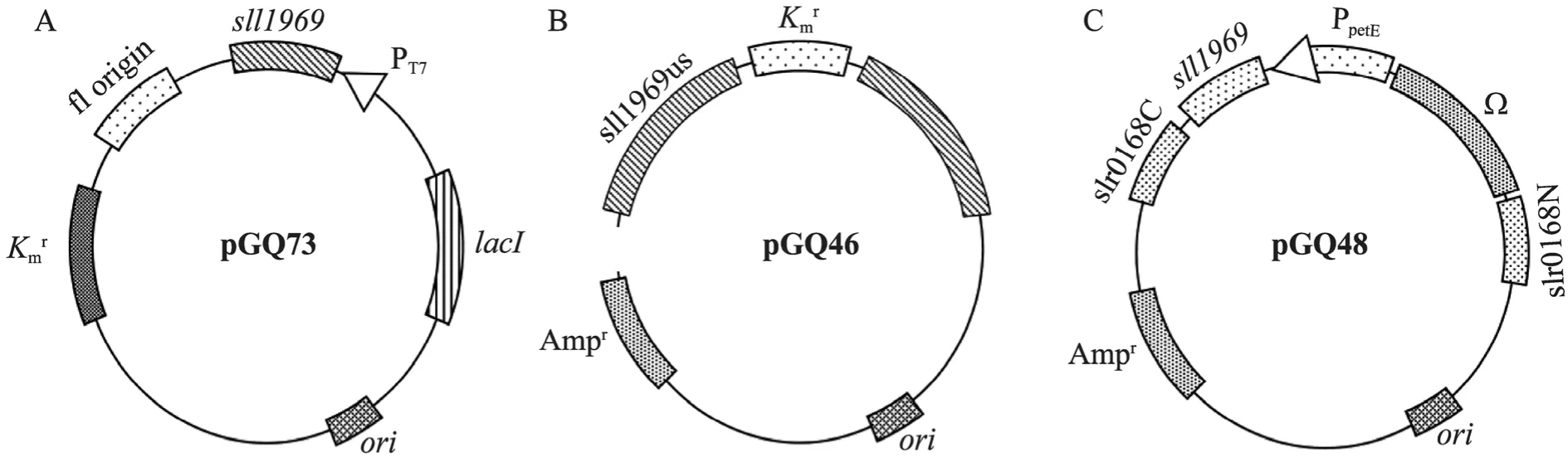
图1 本研究构建的质粒图Fig. 1 Maps of the plasmids. (A) pGQ73. (B) pGQ46. (C) pGQ48.
因为蓝细菌细胞包含多拷贝染色体[22],突变株是否分离完全,需要经过基因组 PCR鉴定(图3)。过表达菌株基因组通过两个PCR反应确定基因插入了正确的位置且整合是完全的 (图3A)。第一个反应使用插入位点的引物 0168-2和插入基因的引物sll1969bglIIs来证实基因插入了正确的位点。第二个反应使用插入位点的引物0168-1和0168-2来检测野生型被取代完全。sll1969敲除突变株的基因型检测,使用敲除位点外围引物sll1969bglIIs和 Sll1969xhoIcas来确定插入是否完全(图 3B)。PCR结果显示该突变株构建分离完全。

图2 SDS-PAGE (A)和Western blotting (B) 检测Sll1969蛋白过表达及纯化及温度对酶活力的影响(C)Fig. 2 SDS-PAGE (A) and Western blotting (B) analysis of Sll1969 and effect of temperature on the activity of Sll1969 (C). (A) M: protein marker; 1: cell lysate of E. coli BL21 containing pGQ73; 2: flow through material; 3:effluent of binding buffer with 20 mmol/L imidazole; 4: effluent of binding buffer with 40 mmol/L imidazole; 5:effluent of binding buffer with 60 mmol/L imidazole; 6: effluent of binding buffer with 100 mmol/L imidazole. (B)Western blotting of purified Sll1969. (C) Effect of temperature on the activity of Sll1969.
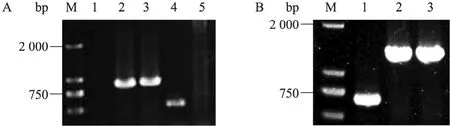
图3 双突变株GQ12和GQ13基因型检测结果Fig. 3 PCR analysis of the genotype of GQ12 (A) and GQ13 (B). (A) M: DNA marker (DL2000 DNA Ladder Marker); 1: genomic DNA of wild type was amplified by primers 0168-2 and sll1969bglIIs (control); 2: genomic DNA of GQ12 was amplified by the same primers as lane 1; 3: plasmid pGQ48 was amplified by the same primer as lane 1(control); 4: genomic DNA of wild-type was amplified by primers 0168-1 and 0168-2 (control); 5: genomic DNA of GQ12 was amplified by the same primers as lane 4. (B) M: DNA marker (DL2000 DNA Ladder Marker); 1: genomic DNA of wild type was amplified by primers sll1969bglIIs and Sll1969xhoIcas (control); 2: genomic DNA of GQ13 was amplified by the same primers as lane 1; 3: plasmid pGQ46 was amplified by the same primer as lane 1 (control).
2.3 突变株自由脂肪酸组成表明 Sll1969不是集胞藻PCC6803细胞中唯一的脂肪酶
我们检测和分析了sll1969过表达突变株GQ12和sll1969敲除突变株GQ13的自由脂肪酸组成和含量的变化 (图4)。由于体外酶学性质分析实验表明该脂肪酶的最适反应温度为55 ℃,因此在检测sll1969过表达对脂肪酸代谢的影响时,将突变株GQ8和GQ12置于42 ℃热激后进行脂肪酸测定。
在42 ℃热激条件下,GQ12和GQ8的自由脂 肪 酸 产 量 分 别 是 (9.2±1.5) mg/(L·OD730)和(6.6±0.5) mg/(L·OD730) (图 4A)。其中过表达sll1969使得脂肪酸含量提高了 40%左右,这表明在这种培养条件下sll1969过表达确实能够提高膜脂向自由脂肪酸的转化。
而30 ℃培养的sll1969敲除突变株GQ13和出发菌株 GQ8的自由脂肪酸含量分别为(3.82±0.48) mg/(L·OD730)和(5.09±0.14) mg/(L·OD730)(图4B),sll1969敲除以后自由脂肪酸的含量下降了33%左右。这也同样印证了上面的结果,sll1969的表达量与细胞的自由脂肪酸含量呈正相关。这与脂肪酶的生理功能是相符合的。值得注意的是,不饱和脂肪酸在两个菌株中产量差别不太明显,GQ13 中的含量是(1.33±0.34) mg/(L·OD730),GQ8中的含量是(1.88±0.51) mg/(L·OD730)。Sll1969 敲除以后不饱和脂肪酸并没有特别明显减少。
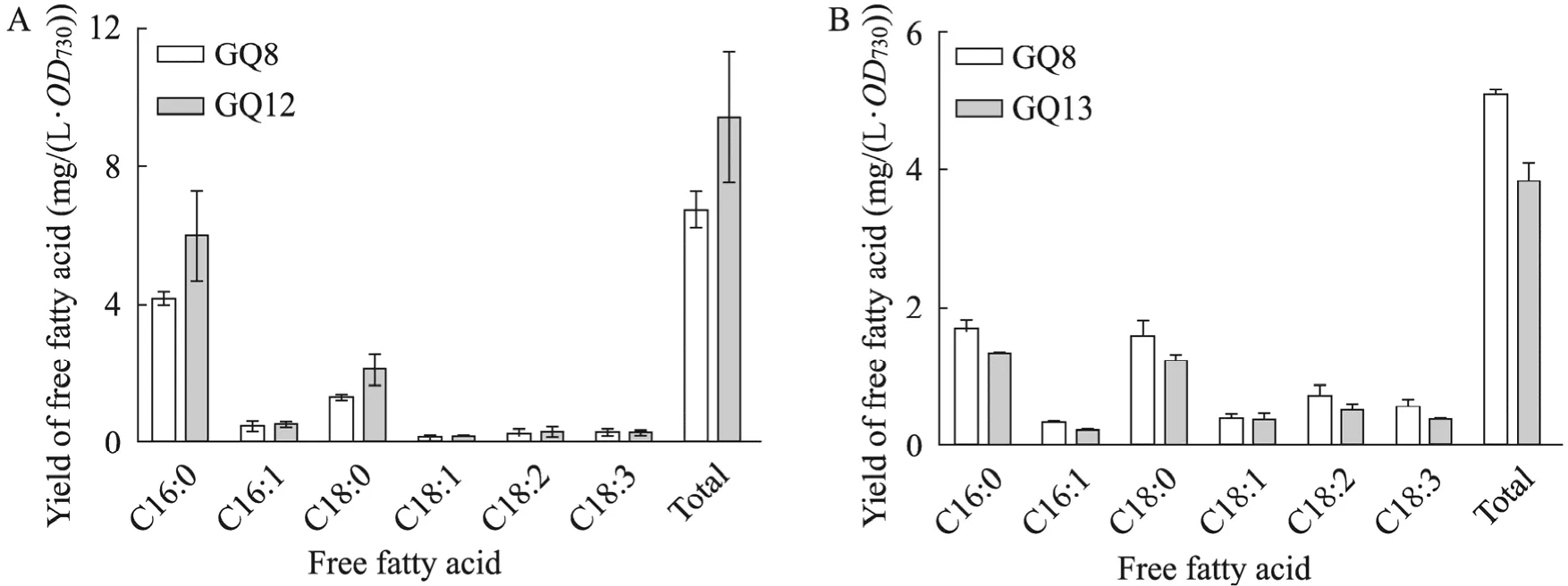
图4 藻株GQ8与sll1969过表达突变株 (GQ12) (A) 和sll1969敲除突变株 (GQ13) (B) 自由脂肪酸产量Fig. 4 Production of free fatty acids of GQ8, GQ12 (A) and GQ13 (B).
3 讨论
生物细胞内自由脂肪酸的来源,一是通过硫酯酶水解脂肪酸生物合成的产物脂酰ACP生成;二是通过脂肪酶水解细胞膜脂而生成。脂酰ACP合酶 (Aas) 敲除突变株自由脂肪酸分析结果显示,相对于野生型,该突变株不饱和自由脂肪酸含量上升[11,16]。而集胞藻PCC6803的脂酰脱饱和酶 (Acyl-lipid desaturase) 的底物为细胞膜脂[23],而非脂酰 ACP;细胞内的脂肪酸侧链只有先整合到细胞膜脂才能被脱饱和。因此,脂酰ACP合酶敲除突变株中不饱和自由脂肪酸含量上升的结果表明,集胞藻 PCC6803细胞内的自由脂肪酸主要来源于细胞膜脂,而不饱和自由脂肪酸的来源一定是细胞膜脂。而 Kaczmarzyk等通过向集胞藻 PCC6803培养液中添加放射性标记的乙酸的同位素示踪实验,发现放射性标记首先出现在细胞膜脂上,然后出现在自由脂肪酸中,这一结果进一步表明细胞内自由脂肪酸来源于膜脂的水解[11]。
通过序列同源比对,集胞藻 PCC6803基因组中的脂肪酶编码基因只有一个,即sll1969基因。本研究中sll1969编码的蛋白的酶学性质分析结果证明这个基因编码蛋白为脂肪酶,其最适反应温度为55 ℃。在此基础上,进一步考察了sll1969突变菌株中脂肪酸含量和组成,分析了sll1969在脂肪酸代谢中的作用。不同菌株脂肪酸含量和组成对比表明sll1969基因表达量与自由脂肪酸产量呈正相关。但是,sll1969基因敲除突变株GQ13中仍然存在自由脂肪酸,特别是不饱和自由脂肪酸的存在,也提示Sll1969不是集胞藻PCC6803细胞中唯一的脂肪酶。而对sll1969基因功能的研究,有助于更清晰地了解蓝细菌脂肪酸代谢以及利用光合蓝细菌平台生产脂肪酸族生物燃料。
[1]Lu XF, Vora H, Khosla C. Overproduction of free fatty acids inE.coli: implications for biodiesel production. Metab Eng, 2008, 10(6): 333−339.
[2]Steen EJ, Kang YS, Bokinsky G, et al. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature, 2010,463(7280): 559−562.
[3]Zhang FZ, Carothers JM, Keasling JD. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol, 2012, 30(4) 354−359.
[4]Lu XF. A perspective: Photosynthetic production of fatty acid-based biofuels in genetically engineered cyanobacteria. Biotechnol Adv, 2010, 28(6):742−746.
[5]Robertson DE, Jacobson SA, Morgan F, et al. A new dawn for industrial photosynthesis. Photosynth Res, 2011, 107(3): 269−277.
[6]Schirmer A, Rude MA, Li XZ, et al. Microbial biosynthesis of alkanes. Science, 2010, 329(5991):559−562.
[7]Tan XM, Yao L, Gao QQ, et al. Photosynthesis driven conversion of carbon dioxide to fatty alcohols and hydrocarbons in cyanobacteria. Metab Eng, 2011, 13(2): 169−176.
[8]Liu XY, Sheng J, Curtiss R III. Fatty acid production in genetically modified cyanobacteria.Proc Natl Acad Sci USA, 2011, 108(17): 6899−6904.
[9]Liu XY, Fallon S, Sheng J, et al.CO2-limitation-inducible green recovery of fatty acids from cyanobacterial biomass. Proc Natl Acad Sci USA, 2011, 108(17): 6905−69058.
[10]Ruffing AM, Jones HDT. Physiological effects of free fatty acid production in genetically engineeredSynechococcus elongatusPCC 7942. Biotechnol Bioeng, 2012, 109(9): 2190−2199
[11]Kaczmarzyk D, Fulda M. Fatty acid activation in cyanobacteria mediated by acyl-acyl carrier protein synthetase enables fatty acid recycling. Plant Physiol, 2010, 152(3): 1598−1610.
[12]von Berlepsch S, Kunz HH, Brodesser S, et al. The Acyl-acyl carrier protein synthetase fromSynechocystissp. PCC6803 mediates fatty acid import. Plant Physiol, 2012, 159(2): 606−617
[13]Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. CSHL Press, 2001.
[14]Elhai J, Wolk CP. A versatile class of positive-selection vectors based on the nonviability of palindrome-containing plasmids that allows cloning into long polylinkers. Gene, 1988, 68(1):119−138.
[15]Williams JGK. Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods inSynechocystis6803.Method Enzymol, 1988, 167: 766−778.
[16]Gao QQ, Wang WH, Zhao H, et al. Effects of fatty acid activation on photosynthetic production of fatty acid-based biofuels inSynechocystissp.PCC6803. Biotechnol Biofuels, 2012, 5(1): 17.
[17]Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 1976, 72(1/2): 248−254.
[18]Nguyen LN, Dao TT, Živković T, et al. Enzymatic properties and expression patterns of five extracellular lipases ofFusarium graminearum in vitro. Enzyme Microb Technol, 2010, 46(6):479−486.
[19]Rippka R, Deruelles J, Waterbury JB, et al. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology, 1979,111(1): 1−61.
[20]Guan WN, Zhao H, Lu XF, et al. Quantitative analysis of fatty-acid-based biofuels produced by wild-type and genetically engineered cyanobacteria by gas chromatography-mass spectrometry. J Chromatogr A, 2011, 1218(45): 8289−8293.
[21]Gao H, Tang Q, Xu XD. Construction of copper-induced gene expression platform inSynechocystissp. PCC6803. Acta Hydrobiol Sin,2007, 31(2): 240−244.
高宏, 唐蜻, 徐旭东. 集胞藻 PCC6803铜离子诱导表达平台的构建. 水生生物学报, 2007, 31(2):240−244.
[22]Griese M, Lange C, Soppa J. Ploidy in cyanobacteria. FEMS Microbiol Lett, 2011, 323(2):124−131.
[23]Murata N, Wada H, Gombos Z. Modes of fatty-acid desaturation in cyanobacteria. Plant Cell Physiol,1992, 33(7): 933−941.
May 4, 2012; Accepted: May 17, 2012
Xuefeng Lü. Tel/Fax: +86-532-80662629; E-mail: lvxf@qibebt.ac.cn
国家自然科学基金 (No. 30970048) 资助。
Characterization of a key gene in membrane lipid cycle inSynechocystissp. PCC6803
Qianqian Gao1,2, Xiaoming Tan1, and Xuefeng Lü1,3
1Key Laboratory of Biofuels,Qingdao Institute of Bioenergy and Bioprocess Technology,Chinese Academy of Sciences,Qingdao266101,Shandong,China
2Graduate University of Chinese Academy of Sciences,Beijing100049,China
3Shandong Provincial Key Laboratory of Energy Genetics,Qingdao266101,Shandong,China
Free fatty acid profiles of wild type and fatty acyl-ACP synthase deletion mutant strain ofSynechocystissp.PCC6803 indicated that one origin of these fatty acids is the process of lipid remodeling or lipid degradation. Lipase is the key enzyme involved in this process. The genesll1969is the sole gene encodes a putative lipase inSynechocystissp.PCC6803. To identify the function of this gene and its role in fatty acid metabolism, we cloned thesll1969from genomic DNA, overexpressed it inEscherichia coliBL21 (DE3) using pET expression system and purified this recombinant enzyme with Nickel-nitrilotriacetic acid affinity chromatography. The enzyme activity was assayed by spectrophotometric with p-nitro-phenylbutyrate as substrate. TheKmandkcatof the enzyme is (1.16±0.01) mmol/L and (332.8±10.0)/min,respectively toward p-nitro-phenylbutyrate at 30oC. The optimal temperature of the enzyme is 55oC. To investigate the biological role of Sll1969 in fatty acid metabolism in cyanobacteria, we constructedsll1969deletion and overexpression mutant strains in the background of fatty acyl-ACP synthase deletion mutant ofSynechocystissp. PCC6803. The analyses of the content of free fatty acids in different mutant strains showed that the contents of Sll1969 and free fatty acid are positively correlated. The free fatty acid profiles of thesll1969mutant strains suggested this enzyme is not the sole enzyme for degrading lipid inSynechocystissp. PCC6803.
lipase,sll1969, p-nitro-phenylbutyrate, fatty acid
Supported by: National Natural Science Foundation of China (No. 30970048).
