脊髓小脑性共济失调与多系统萎缩临床鉴别诊断
2012-06-07吴媛媛张佩兰
吴媛媛 张佩兰
脊髓小脑共济失调(spinocerebellar ataxias,SCAs)是一大类以累及人类中枢神经系统为主的进行性神经系统变性病,是一种单基因遗传病,多为常染色体显性遗传,临床上以进行性小脑性共济失调为主要症状。按照基因型分类,迄今为止共发现SCA 1~26等若干亚型。SCA和MSA患者在临床表现和核磁影像学表现相近,尤其是遗传史不明的SCA患者同OPCA患者的鉴别非常困难。本文收集我院2005年12月至2010年8月神经内科SCA及MSA患者临床资料及主要辅助检查进行回顾性分析,鉴别诊断提供依据。
1 资料与方法
1.1 一般资料 病例组A来源门诊临床初步诊断的SCA家系患者。收集9个家系共计患病22例,均为汉族。目前共发病患者22例,男10例,女12例;发病年龄18~48岁,平均年龄31.30岁;女性平均发病年龄29.00岁,男性平均发病年龄33.60岁;病程平均9.10年。病例组B来源于门诊临床初步诊断的多系统萎缩(MSA)患者10例,女6例,男4例;发病年龄40~62岁,平均年龄51.24岁;病程平均5.2年。2组发病年龄比较差异有统计学意义(P<0.05)。
1.2 方法 SCA组参照国际统一SCA临床症状诊断Harding标准,MSA组参照标准美国学者Gilman于1999年提出的诊断标准,进行“可能的诊断”或“很可能的诊断”。除外代谢障碍所致脑病的可能:对所有患者均完善各项辅助检查,除外已知生化异常所致共济失调的可能,如维生素E缺乏共济失调、低β蛋白血症、线粒体脑肌病、肝豆状核变性等。
2 结果
2.1 家系调查 SCA组遗传形式9个家系均符合染色体显性遗传。遗传无男女差别。其中张氏家系连续遗传4代,患病者10例,男女比例5∶5,符合常染色体显性遗传。MSA组,均为散发。个别MSA患者追问病史回忆述上一代老年期偶出现类似表现。见图1。
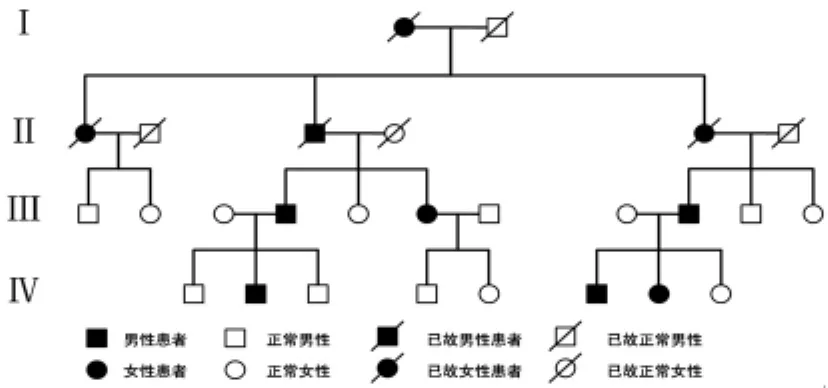
图1 张氏家系遗传图谱
2.2 临床表现 SCA组:共同特征:所有发病者均有平衡障碍、共济失调,构音障碍。最早出现的临床症状是走路不稳,言语障碍可与走路不稳同时出现,也可推后出现,上述症状随病程的发展而逐渐加重,临床症状随病情进展加重明显。发病SCA患者的临床特征:肢体共济失调和构音障碍患者9例;病理征阳性患者4例;肌张力增高患者3例;饮水呛咳患者2例;二便障碍患者3例;偶有肢体抽搐患者1例;听力患者障碍2例;眼震患者2例;阳痿患者1例;无1例出现感觉障碍。MRA组:症状主要分为小脑共济失调,自主神经功能障碍,帕金森样症状3组。首发行走困难或僵硬2例;首发症状震颤6例;肌张力增高患者3例;语言障碍患者5例;二便障碍患者3例;体位性低血压症状患者5例;眼震患者2例;姿势反射异常患者1例。
2.3 MRI表现 共同表现:19例均行MRI检查,发现有不同程度的小脑或脑干、大脑皮层萎缩,MRI表现:(1)SCA组:13例均可见小脑皮层脑沟变深变宽,脑回变薄,小脑蚓部萎缩、消失;桥脑、延髓体积缩小。6例合并额叶、顶叶等大脑皮层萎缩,5例均可见小脑皮层脑沟变深、变宽或小脑蚓部萎缩。1例还合并脑干轻度萎缩。见图2。(2)MSA组:2例壳核外侧缘高信号;5例桥脑萎缩;8例桥脑基底部十字征;4例第四脑室扩大;2例小脑萎缩(半球>蚓部)沟裂增宽加深,小脑小叶变细变直;6例皮层萎缩(额顶叶为著)。2例脑干变细,桥脑腹侧面萎缩变平。见图3。
2.4 MSA患者进行了MRS检查 1例丘脑 NAA及小脑半球NAA下降,CR升高CHO下降,右基底节右内囊后肢与双丘脑NAA下降CR升高,CHO下降。
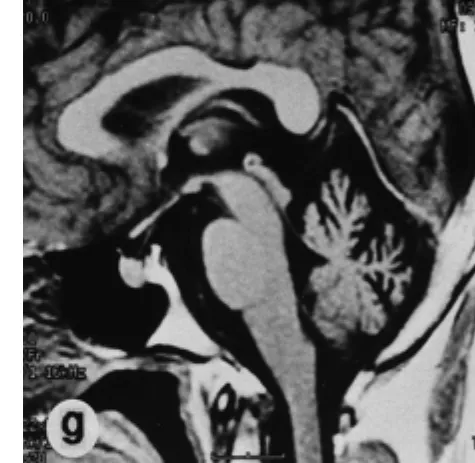
图2 SCA患者小脑上蚓部明显萎缩
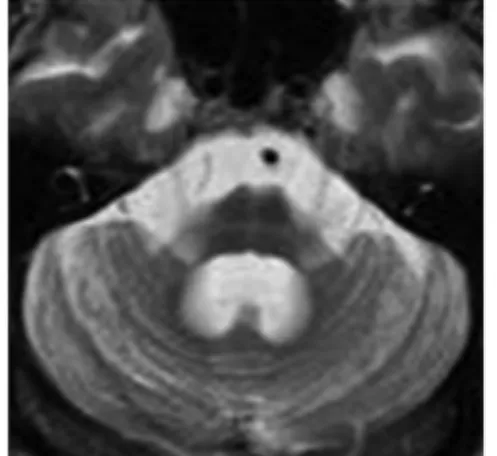
图3 MSA患者十字征
2.5 3例MSA患者行肛门括约肌电生理检查 2例均有肛门括约肌肌神经源损害。
3 讨论
SCAs是一种常见的神经系统遗传病,在神经系统遗传病中发病率位居第3位,同时也是目前研究较广和较深的遗传性疾病。由于该病的遗传异质性和基因突变的多态性,病变主要侵犯小脑、脊髓和脑干。SCA和MSA患者在临床症状重叠,核磁影像学表现相近,SCA诊断时需要排除MSA等有小脑脑干萎缩表现的神经系统变性病,且SCA患者同遗传家族史不详的以小脑症状为主的MSA患者的鉴别非常困难。临床诊断易造成误诊。本研究表明:SCA家族发病符合常染色体显性遗传规律,发病年龄越晚,临床症状以及病情进展相对较轻、病程相对较长。发病与性别无关,男女均可患病。相比MSA,SCA患者发病年龄相对提前。MSA患者以中年发病为主,同样发病与性别无关,鲜见遗传史,多为散发病例。家族史的调查对于明确二者的诊断有重要意义。
临床症状鉴别:1900年Dejerine和Thomas首先提出“OPCA”的概念,以描述1例共济失调患者的病理特征,即脑桥基底部、下橄榄体和小脑中脚变性。MSA-C型即以小脑症状为主要表现即属于这一范畴。与SCA临床症状相比。二者相比,走路不稳,构音障碍是SCA更为常见的症状,这与雷晶等[1]的结果一致。对于临床症状的二者早期鉴别来说,自主神经功能障碍,和以帕金森样症状为主要表现的MSA可以对二者进行区别。王宪玲等[2]在观察10例MSA患者中发现8例出现自主神经症状,这一比例远高于其他神经系统变性病出现自主神经症状的概率。本组研究患者中首发行走困难或僵硬2例;姿势反射异常患者1例;体位性低血压症状患者5例,这些特殊鉴别症状在SCA组患者中并未发现。
核磁影像上鉴别:SCA的病理损害主要是小脑蒲肯野氏细胞、脑干以及脊髓神经纤维脱髓鞘改变,因此在影像学的表现也是以上述部位的脑萎缩为主,Giuffvida等[3]对20例SCA2型进行核磁检查发现opca是SCA2的一种典型的类型,20例SCA2中有12例存在幕上结构的萎缩。并且萎缩程度和病程相关。Adachi等[4]发现 T2像脑桥基底部中线高密度影是SCA1的特征性表现之一。Schulz等[5]通过3 d容积核磁技术发现,sca3小脑半球萎缩与其他类型相比并不明显,sca6的小脑萎缩部位集中在灰质。Lukas等[6]发现脊髓高位萎缩是sca3的除小脑萎缩萎缩以外的特征表现。Murata等[7]认为小脑传入和传出纤维束、苍白球及额、颞叶萎缩是SCA3/MJD病的特征性改变。本研究发现SCA患者核磁主要特征以小脑蚓部及半球的萎缩为主,萎缩犹以小脑蚓部为重,脑干的萎缩不明显,桥脑腹侧向前隆起的弧度存在。结合本次研究结果考虑为SCA2,3两型结果可能性大,明确需要进一步基因诊断。
MSA病理损伤机制可能是脑桥核及脑桥横向纤维变性,胶质增生含水量增加,而由齿状核发出构成小脑上脚的纤维和锥体束未受损害有关。核磁特征:提示幕下结构异常表现为脑干形态变细,小脑体积变小,沟裂增宽加深,半球小叶变细变直呈枯树枝状。脑池及脑室扩大,脑桥小脑中脚以及小脑T2WI对称性高信号。基底节区异常表现为壳核萎缩,壳核背外缘T2WI低信号或外侧缘缝隙样高信号[8]。Adachi等[9]通过对12例脊髓小脑萎缩型病人观察得出,通过核磁检查可见DWI序列可以作为观察脑桥横向纤维的技术手段,opca有脑桥,小脑蚓部的异常高信号,从而为区别opca与SCA各型提供线索。张雪宁等[10]对20例opca进行核磁检查发现4例伴有壳核背外侧缘信号减低。本次研究中发现2例壳核外侧缘高信号。额顶叶萎缩考虑患者有相应的智能减退。
“十字征”对于OPCA与SCA的鉴别意义存在争议,有研究者发现SCAs病例也可有这种影像学特征,而另一些研究则显示仅在OPCA出现“十字征”。顾卫红等[11]认为存在这种争议可能的原因为研究者所收集的病例的病程不同,影像学检查的分辨率存在差异及scA具有高度的临床异质性等,本次研究中8例MSA患者出现十字征,“十字征”作为OPCA的特征性影像学改变,还需要大量临床影像学资料进一步证实。
MRS质谱分析也是辅助鉴别的一项重要技术,对于MSA患者,有资料显示脑桥基底部质子波谱分析N-乙酰天冬氨酸/肌酐(NAA/Cr)值显著降低,本研究由于条件所限仅对1例MSA患者进行了MRS检查:丘脑NAA及小脑半球 NAA下降,CR升高CHO下降,右基底节右内囊后肢与双丘脑 NAA下降CR升高,CHO下降。结果对诊断有重要提示作用。
MSA患者比较突出的临床特征伴有自主神经功能障碍,患者骶髓前角细胞(Onuf核)的变性导致了肛门和尿道括约肌的失神经支配和再支配,表现为运动单位电位的改变。因此肛门括约肌EMG检查对MSA的诊断,尤其在早期与其他神经系统变性病的鉴别诊断具有很大的临床价值。
综上所述,对于SCA和MSA,尽管临床症状重叠,仍可找到鉴别点,详查家族病史,综合考虑多种临床症状和神经查体,并结合辅助检查,尤其是核磁影像学检查,有条件的可以行基因诊断,可提高两者鉴别诊断的准确性。
1 雷晶,马建华,张小宁.8例脊髓小脑共济失调2型、3型患者的临床表型及MRI表现分析.新疆医科大学学报,2007,30:1245-1248.
2 王宪玲,贾建平.多系统萎缩与帕金森病的早期鉴别诊断.中风与神经疾病杂志,2003,20:27-29.
3 Giuffvida S,Saponara R,Restivo DA,et al.Supratentorial atrophy in spinocerebellar ataxia type 2:MRI study of 20 patients.J Neurol,1999,246:383-388.
4 Adachi M,Kawanami T,Ohshima T,et al.Characteristic signal changes in the pontine base on T2-and multishot diffusion-weighted images in spinocerebellar ataxia type 1.Neuroradiology,2006,48:8-13.
5 Schulz JB,Borkert J,Wolf S,et al.Visualization,quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1,3 and 6.NeuroImage,2010,49:158-168.
6 Lukas C,Hahn HK,Bellenberg B,et al.Spinal cord atrophy in spinocerebellar ataxia type 3 and 6:Impact on clinical disability.J Neurol,2008:255:1244-1249.
7 Murata Y,Yamaguchi S,Kawakami H,et al.Characteristic magnetic resonance imaging findings in Machado-Joseph disease.Arch Neurol,1998,55:33-37.
8 吴江,贾建平,崔丽英主编.神经病学.第2版.北京:人民卫生出版社,2010.151-193.
9 Adachi M,Hosoya T,Yamaquchi K,et al.Diffusion and T2-weight MRI of the transverse pontine fibres in spinocerebellar degeneration.Neuroradiology,2000,42:803-809.
10 张雪宁,廉宗徽.橄榄桥小脑萎缩的临床与磁共振诊断.天津医药,1996,24:690-691.
11 顾卫红,王国相.橄榄脑桥小脑萎缩、脊髓小脑共济失调和多系统萎缩小脑型的诊断和鉴别.中华神经科杂志,2008,41:135-137.
