血管紧张素转换酶抑制剂和血管紧张素Ⅱ受体拮抗剂单药治疗及联合应用对慢性肾脏病患者肾素-血管紧张素系统表达的影响
2012-05-10章晓燕於佳炜刘春凤钟一红丁小强
章晓燕 於佳炜 刘春凤 滕 杰 钟一红 丁小强
肾脏具有不依赖于循环肾素-血管紧张素系统(renin angiotensin system,RAS)的局部RAS系统,肾脏局部RAS过度兴奋是慢性肾脏病(chronic kidney disease,CKD)进展的重要因素,针对RAS的干预药物包括血管紧张素转换酶抑制剂(angiotensin converting enzyme inhibitor,ACEI)、血管紧张素Ⅱ受体拮抗剂(angiotensinⅡreceptor blocker,ARB)和肾素抑制剂等,作为治疗CKD的重要工具,其疗效得到许多循证医学试验的验证[1]。但目前关于RAS抑制剂对人类循环RAS活性影响的研究很少,对CKD患者肾脏RAS表达影响的研究更少,而且循环RAS的活性并不能直接反映肾脏RAS的表达[2]。所以RAS抑制剂虽已被广泛运用于治疗CKD,但仍缺乏其对CKD患者肾脏RAS活性影响的直接数据。目前已有一些动物研究和一项临床研究提示尿血管紧张素原(angiotensinogen,AGT)水平与肾脏血管紧张素Ⅱ(angiotensinⅡ,AngⅡ)含量或免疫组化染色强度呈正相关,故尿AGT被认为是反映肾脏AngⅡ活性的无创指标[3-5]。本研究旨在以尿AGT作为肾脏AngⅡ活性指标,探讨ACEI和ARB单药及联合应用对CKD患者循环和肾脏RAS活性的影响,并比较这三种治疗方法之间的差异。
对象和方法
研究对象 2009年4月至2010年1月复旦大学附属中山医院肾内科门诊随访的24例CKD患者。纳入标准:年龄16~70岁;2次蛋白尿定量均值在0.15~1.5 g/24h;估算肾小球滤过率(estimated glomerular filtration rate,eGFR)≥ 30 ml/(min·1.73m2);取得患者同意并签署知情同意书。
排除标准:存在系统性红斑狼疮、过敏性紫癜、慢性肝脏疾病、肿瘤、糖尿病等继发性因素;肥胖、感染、酗酒或吸毒;口服避孕药、妊娠和哺乳期妇女;肾血管性高血压;梗阻性肾病;血压>160/100 mmHg;3月内曾使用ARB或ACEI、β受体阻滞剂、可乐定、利尿剂;既往1年内曾使用激素、非甾体类消炎药或免疫抑制剂;有肾活检禁忌证;体重过重或过轻者。
分组和用药 采用前瞻性随机对照设计,入选患者按时间顺序编号,根据数字随机表分为贝那普利组(ACEI组)、缬沙坦组(ARB组)、贝那普利和缬沙坦联合治疗组(ACEI+ARB组)。ACEI组贝那普利起始剂量10 mg/d,2周后加至20 mg/d;ARB组缬沙坦起始剂量80 mg/d,2周后加至160 mg/d;ACEI+ARB组从贝那普利10 mg/d开始,2周后加用缬沙坦80 mg/d。治疗过程中如患者出现低血压,则不再加量,如发生不能耐受的干咳,则剔除,并由即将入组的患者替补,总疗程8周。研究期间不使用糖皮质激素、免疫抑制剂、β受体阻滞剂、利尿剂、甘草制剂及贝那普利、缬沙坦以外的其他ACEI/ARB。目标血压<130/80 mmHg,合并高血压的患者在计划用药后血压仍无法达标者优先使用氨氯地平(最大剂量10 mg/d)控制血压,仍无法达标者允许使用胍乙啶等对RAS活性影响较小的药物。研究期间所有患者采用低盐饮食(氯化钠<5 g/d),并以尿钠排泄率评价饮食依从性,目标尿钠80~100 mmol/24h。以治疗8周后24h尿蛋白定量下降>30%为主要终点。研究方案经复旦大学附属中山医院伦理委员会批准。
观察指标 研究对象每2周随访1次,记录以下临床指标和血、尿RAS组分活性。
临床指标 包括性别、年龄、身高、体重、血压、尿常规、肾功能、电解质、24h尿蛋白定量、尿钠。并根据MDRD简化公式计算eGFR[eGFR=175×血清肌酐(SCr)-1.154×年龄-0.203×(0.741,亚裔)×(0.742,女性)][6]。
标本留取 血标本留取:采肘静脉血4 ml分别冰水浴冷却的EDTA抗凝管和血清管,1 000 r/min 4℃离心5 min,分离血浆和血清,分装后-20℃冰箱保存,用于测定血浆肾素活性(plasma renin activity,PRA)、AGT、Ang Ⅱ和醛固酮。
尿标本留取:新鲜晨尿 10ml,冰水浴冷却,1 000 r/min 4℃离心5 min,分离上清液,分装后-20℃冰箱保存,用于测定尿 AGT、AngⅡ和醛固酮。
血、尿RAS组分的检测 采用人血管紧张素Ⅰ放射免疫分析试剂盒(北京原子高科股份有限公司)、人血管紧张素Ⅱ放射免疫分析试剂盒(北京原子高科股份有限公司)、人总血管紧张素原ELISA试剂盒(日本Immuno-Biological Laboratories)和人醛固酮 ELISA 试 剂 盒 (日 本 Immuno-Biological Laboratories)测定PRA、AGT、AngⅡ和醛固酮。
统计学处理 所有数据均采用SPSS 17.0统计软件分析处理。计量资料以平均值±标准差表示,计数资料以绝对例数或百分比(%)表示。各组治疗前后的数据比较采用配对t检验,组间数据比较采用卡方检验或t检验。P<0.05为差异有统计学意义。
结 果
患者一般情况 24例患者中男性9例,女性15例,年龄43.62±11.47岁(26~65)岁。每组各8例,ACEI组原发病为轻微病变1例、IgA肾病3例、局灶节段增生性肾炎1例、局灶节段硬化性肾炎1例、淀粉样变性1例、1例临床诊断慢性肾小球肾炎;ARB组为轻微病变1例、IgA肾病5例、局灶节段增生性肾炎2例;ACEI+ARB组为轻微病变2例、IgA肾病2例、局灶节段增生性肾炎2例、局灶节段增生性肾炎1例、1例临床诊断慢性肾小球肾炎。三组年龄、性别、身高、体重、血压、SCr、eGFR、24h尿蛋白定量和尿钠等无统计学差异(表1)。
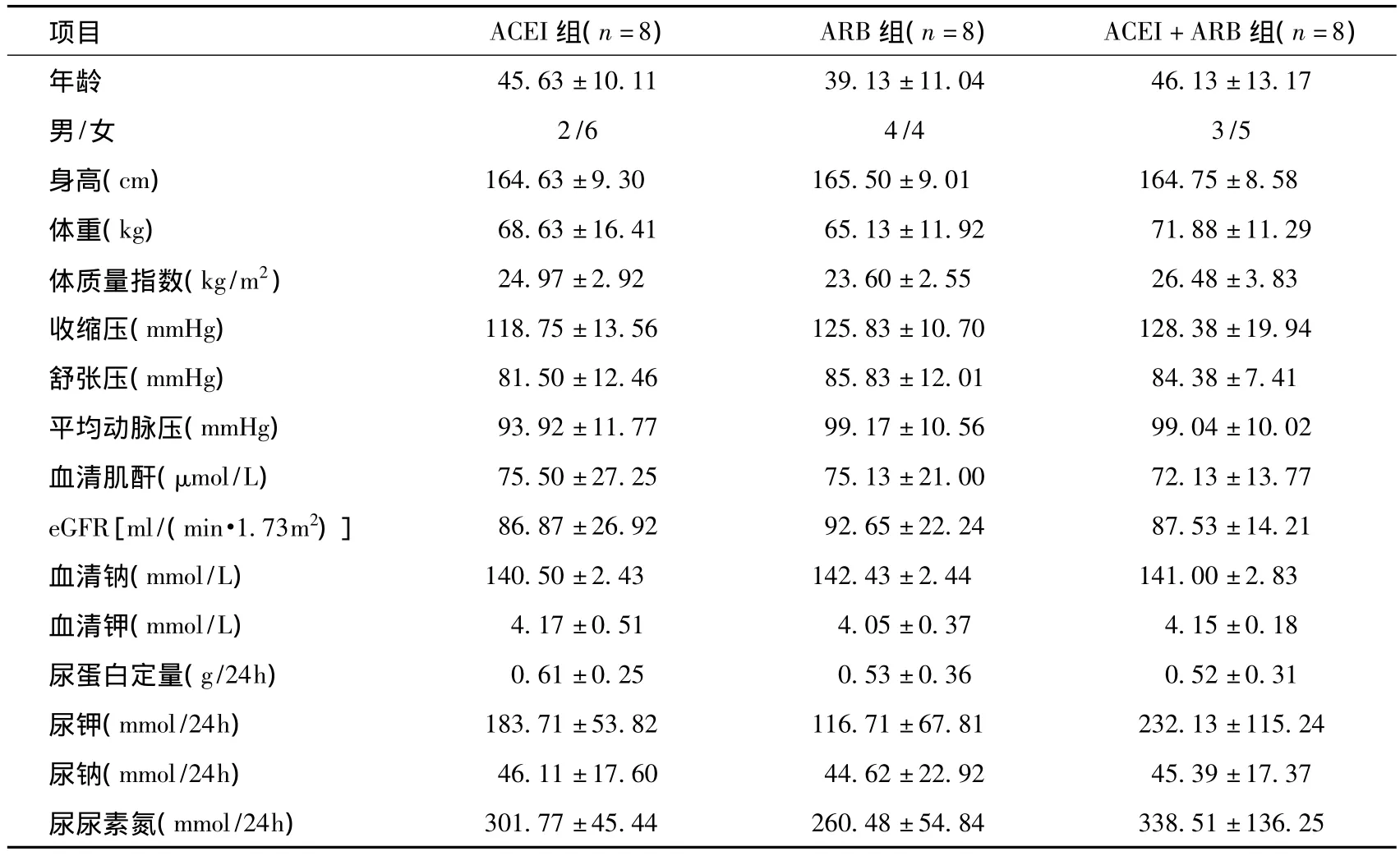
表1 患者基本情况和基线资料
治疗前后临床指标的比较 治疗8周后ACEI组的24h尿蛋白定量低于基线(P<0.05),ARB组的收缩压、舒张压和平均动脉压均低于基线(P<0.05);ACEI+ARB组的临床指标与基线无差异。治疗8周后三组血压、SCr、血钾和尿钠无统计学差异。主要终点事件发生率ACEI组、ARB组和联合治疗组分别为87.5%、12.5%和62.5%,ACEI组显著高于ARB组(P<0.05)(表2、图1)。合并高血压的患者在计划用药后血压达标,无合并使用其他降压药。
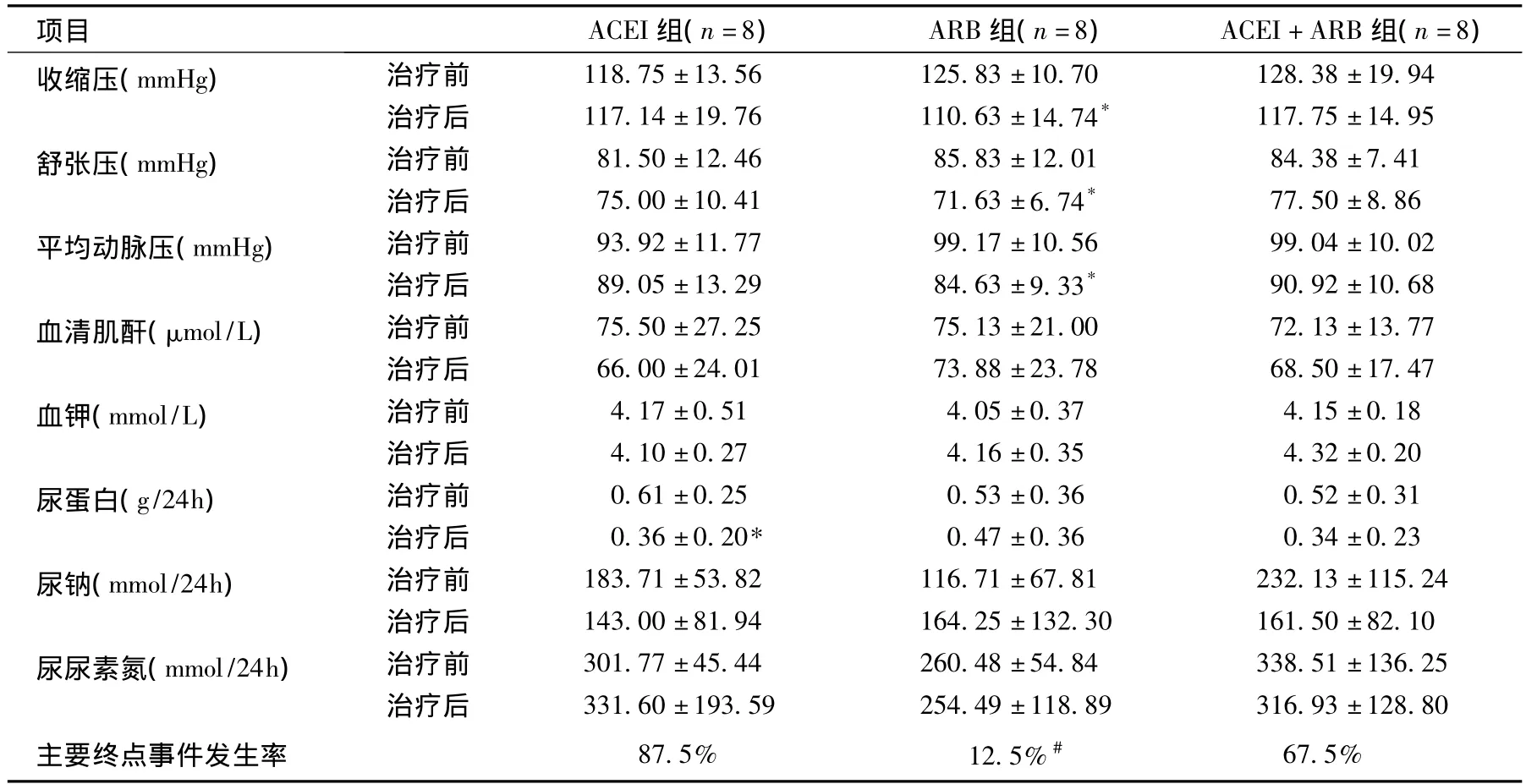
表2 治疗前后临床指标的比较
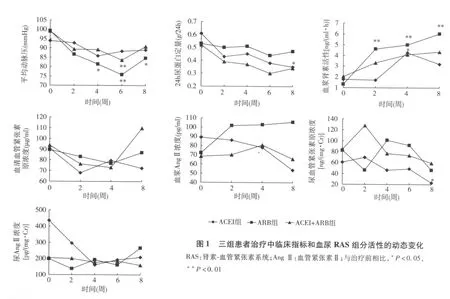
治疗前后血和尿RAS组分活性比较 治疗8周后ACEI组的尿AGT明显下降(P<0.05);ARB组的PRA显著升高(P<0.01)(表3、图1)。治疗前三组PRA和血、尿AGT、AngⅡ和醛固酮活性无统计学差异。治疗8周后,ACEI组 PRA和血浆AngⅡ浓度低于ARB组(P<0.05),ACEI组血清AGT明显低于ACEI+ARB组(P<0.01)(表3)。
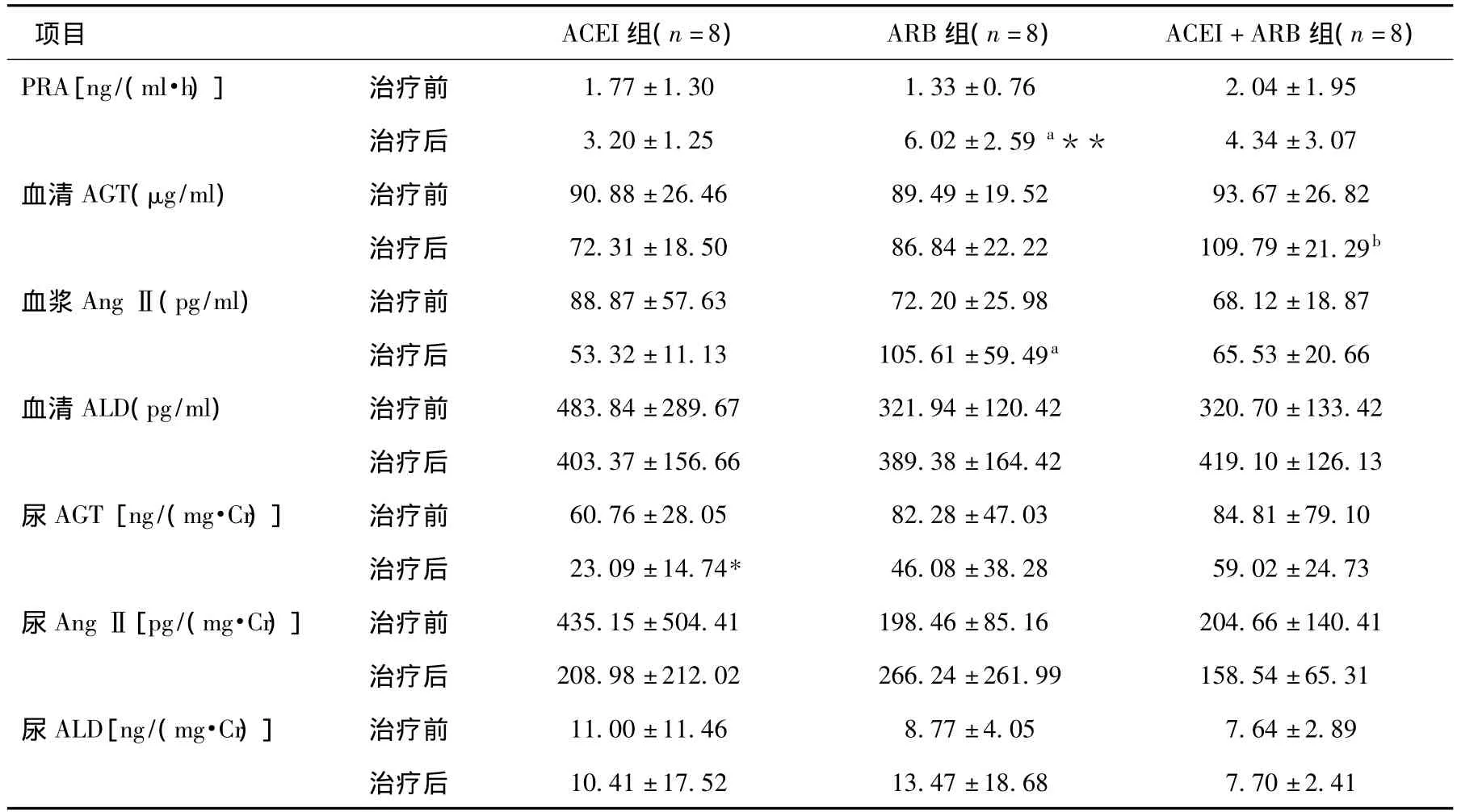
表3 治疗前后血和尿RAS组分活性比较
讨 论
AngⅡ在肾脏的主要作用是通过血管紧张素Ⅱ1型受体(angiotensinⅡ type 1 receptor,AT1R)调节肾小球血流量和肾小管水钠重吸收[7],大量证据表明肾脏高表达AT1R,是RAS的主要靶器官[8]。动物研究提示不同RAS抑制剂对循环和肾脏RAS活性影响不同[9,10]。在自发性高血压大鼠模型中,依那普利和氯沙坦均可降低血浆AGT水平,升高循环和肾脏的肾素活性[9]。坎地沙坦可以升高Dahl盐敏感大鼠血浆AngⅡ,但肾脏AngⅡ活性下降[10]。此外,由于动物研究和一项临床试验提示尿AGT与肾脏AngⅡ活性呈正相关[3-5],有作者以尿AGT作为肾脏局部AngⅡ活性的指标发现,CKD患者使用氯沙坦后血浆AngⅡ水平升高,尿AGT下降,提示氯沙坦可以降低肾脏内AngⅡ活性[5]。但目前尚无不同RAS抑制剂对人类循环和肾脏RAS活性不同影响的直接数据。
本研究发现经8周治疗,ACEI组24h尿蛋白定量、尿AGT均较基线显著下降,提示贝那普利可以显著降低肾脏AngⅡ活性,且可能与其降低蛋白尿的作用相关。ACEI组治疗后AGT、AngⅡ下降,PRA升高,但无统计学意义,变化趋势与既往研究的结果相似[8]。ARB组平均动脉压较治疗前显著下降,PRA显著升高。AngⅡ有升高趋势,血清AGT和尿AGT有下降趋势,差异虽无统计学意义,但变化趋势也与既往的研究结果相似[5,8,9]。联合治疗组PRA略升高,AngⅡ无显著变化,而尿AGT略下降,是ACEI和ARB对循环及肾脏局部RAS活性不同影响的综合结果。但是在ACEI组和ARB组AGT都呈下降趋势的情况下,联合治疗AGT却显著高于基线,是样本量小、随访时间短所引起的偶然性现象,还是另有原因,本研究尚难于确定,需进一步扩大样本量深入研究。
由于AngⅡ分子量小,可自由通过肾小球滤过屏障,故尿液中的AngⅡ一方面来自循环AngⅡ,另一方面来自肾脏局部形成的AngⅡ[10]。ACEI治疗后,循环和肾脏局部AngⅡ活性均趋向于下降,故尿AngⅡ浓度也呈下降趋势,ABR治疗后循环AngⅡ活性趋向于升高,肾脏局部AngⅡ活性趋向于下降,故尿AngⅡ浓度的变化表现为循环和肾脏局部AngⅡ活性变化的综合结果。
本研究结果说明不同RAS抑制剂和治疗方案对循环 RAS不同组分的影响不同。ACEI升高PRA,降低AGT、AngⅡ;ARB升高 PRA、AngⅡ,降低AGT;联合治疗对循环RAS的影响则为两者的综合效应。进一步以尿AGT作为评价肾脏AngⅡ活性的指标发现,不同RAS抑制剂和治疗方案对肾脏局部AngⅡ活性影响相似,表现为尿AGT下降,提示肾脏AngⅡ活性下降。
既往文献报道联合使用小剂量ACEI和ARB可较单一使用大剂量ACEI、ARB更有效地减少Ⅰ型糖尿病患者的微量白蛋白尿[11]和原发性肾小球肾炎患者的显性白蛋白尿[12,13]。本研究发现,治疗8周后ACEI组终点事件发生率高于ARB组,但未发现联合治疗组优于ACEI组和ARB组,三组在血压、SCr、血钾等方面无统计学差异,提示ACEI减少CKD患者蛋白尿的效果略优于ARB。分析可能的原因发现,ACEI组PRA低于ARB组,与既往文献报道相同[10]。最近肾素和前肾素的特异性跨膜受体被克隆[14,15],虽然目前尚未完全明确其功能,但已有结果提示肾素与受体结合不仅可以提高肾素的酶活性[16],而且可对人系膜细胞产生不依赖于AngⅡ的促纤维化作用[17]。肾血管性高血压大鼠肾脏的肾素受体表达上调,并可能与其肾小球损伤相关[18]。有作者进一步使用诱捕多肽阻断前肾素非水解活性相关的结构域,可延缓糖尿病肾病进展,提示肾素的非水解活性可能与糖尿病肾病的进展机制相关[19]。所以,我们推测贝那普利、缬沙坦对PRA的影响不同,并进一步通过肾素受体产生后续效应,这可能是贝那普利更有效减少蛋白尿的原因之一。此外,既往研究提示使用贝那普利2月后尿蛋白排泄率就开始下降[20],而缬沙坦使用3~6月,尿蛋白排泄率才逐渐下降至平稳[21,22],两种药降蛋白尿的起效时间不同也可能是贝那普利减少蛋白尿略优于缬沙坦的原因。
与既往研究不同的是,本研究未发现联合治疗组减少蛋白尿的效果优于ACEI组和ABR组。可能的原因包括:(1)与研究设计的差异有关,既往研究均为交叉对照研究[12,13],不同治疗方式转换之间不设洗脱期,而且每种治疗方式的用药时间比本研究长。也有作者仅发现缬沙坦160 mg/d联合贝那普利5~10 mg/d治疗减少蛋白尿的效果优于缬沙坦160 mg/d,而缬沙坦80 mg/d联合贝那普利5~10 mg/d治疗并不优于缬沙坦160 mg/d[23]。有作者在交叉对照研究中通过对GFR、滤过分数、肾血管阻力、有效肾血浆流量和右旋糖酐清除分数的测定发现,ACEI和ARB联合治疗更好的抗蛋白尿作用可能与ACEI相关的血流动力学效应和肾小球滤过屏障的通透性改善有关[12]。(2)既往研究中CKD患者治疗前的蛋白尿水平都明显高于本研究[12,13]。(3)本研究虽为前瞻性随机对照研究,但仍存在样本量小、随访时间短等缺陷,而且本研究中所采用的贝那普利、缬沙坦剂量为临床常用剂量,亦为可有效避免体位性低血压、高钾血症等RAS抑制剂常见不良反应的“临床安全剂量”,但三组在治疗过程中均未见尿AGT下降至平稳的趋势,提示可能尚存在肾脏局部AngⅡ活性抑制不足的可能。
综上所述,贝那普利可降低CKD患者肾脏局部AngⅡ活性并减少蛋白尿。缬沙坦较贝那普利更显著升高血浆肾素活性和AngⅡ浓度。
1 Brenner BM,Cooper ME,de Zeeuw D,et al.Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy.N Engl J Med,2001,345(12):861 -869.
2 Nishiyama A,Seth DM,Navar LG,et al.Renal interstitial fluid concentrations ofangiotensins Iand IIin anesthetized rats.Hypertension,2002,39(1):129 -134.
3 Kobori H,Harrison-Bernard LM,Navar LG.Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production.Kidney Int,2002,61(2):579 -585.
4 Kobori H,Nishiyama A.Effects of tempol on renal angiotensinogen production in Dahl salt-sensitive rats.Biochem Biophys Res Commun,2004,315(3):746 -750.
5 Yamamoto T,Nakagawa T,Suzuki H,et al.Urinary angiotensinogen as a markerofintrarenalangiotensin IIactivity associated with deterioration of renal function in patients with chronic kidney disease.J Am Soc Nephrol,2007,18(5):1558 -1565.
6 Levey AS,Coresh J,Greene T,et al.Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate.Ann Intern Med,2006,145(4):247 -254.
7 Leong PK,Devillez A,Sandberg MB,et al.Effects of ACE inhibition on proximal tubule sodium transport.Am J Physiol Renal Physiol,2006,290(4):F854 -F863.
8 Velez JC.The importance of the intrarenal renin-angiotensin system.Nat Clin Pract Nephrol,2009,5(2):89 - 100.
9 Menard J,Campbell DJ,Azizi M,et al.Synergistic effects of ACE inhibition and Ang II antagonism on blood pressure,cardiac weight,and renin in spontaneously hypertensive rats.Circulation,1997,96(9):3072-3078.
10 Nishiyama A,Yoshizumi M,Rahman M,et al.Effects of AT1 receptor blockade on renal injury and mitogen-activated protein activity in Dahl salt-sensitive rats.Kidney Int,2004,65(3):972 -981.
11 Jacobsen P,Andersen S,Jensen BR,et al.Additive effect of ACE inhibition and angiotensin II receptor blockade in type I diabetic patients with diabetic nephropathy.J Am Soc Nephrol,2003,14(4):992-999.
12 Campbell R,Sangalli F,Perticucci E,et al.Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies.Kidney Int,2003,63(3):1094 -1103.
13 Rutkowski P,Tylicki L,Renke M,et al.Low-dose dual blockade of the renin-angiotensin system in patients with primary glomerulonephritis.Am J Kidney Dis,2004,43(2):260 -268.
14 Nguyen G,Delarue F,Burcklé C,et al.Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin.J Clin Invest,2002,109(11):1417 -1427.
15 Batenburg WW,KropM,Garrelds IM,et al.Prorenin is the endogenous agonist of the(pro)renin receptor:binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor.J Hypertens,2007,25(12):2441-2453.
16 Nguyen G.The(pro)renin receptor:pathophysiological roles in cardiovascular and renal pathology.Curr Opin Nephrol Hypertens,2007,16(2):129 -133.
17 Huang Y,Wongamorntham S,Kasting J,et al.Renin increases mesangial cell transforming growth factor-β1 and matrix proteins through receptor-mediated,angiotensin II-independent mechanisms.Kidney Int,2006,69(1):105 -113.
18 Krebs C,Hamming I,Sadaghiani S,et al.Antihypertensive therapy upregulates renin and(pro)renin receptor in the clipped kidney of Gold blatt hypertensive rats.Kidney Int,2007,72(6):725 -730.
19 Ichihara A,Hayashi M,Kaneshiro Y,et al.Inhibition of diabetic nephropathy by a decoy peptide corresponding to the“handle”region for nonproteolytic activation of prorenin.J Clin Invest,2004,114(8):1128-1135.
20 Maschio G,Alberti D,Janin G,et al.Effect of the angiotensinconverting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency.The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group.N Engl J Med,1996,334(15):939-45.
21 Li PK,Leung CB,Chow KM,et al.Hong Kong study using valsartan in IgA nephropathy(HKVIN):a double-blind,randomized,placebocontrolled study.Am J Kidney Dis,2006,47(5):751 -760.
22 Shiga Microalbuminuria Reduction Trial(SMART)Group,Uzu T,Sawaguchi M,et al.Reduction of microalbuminuria in patients with type 2 diabetes:the Shiga Microalbuminuria Reduction Trial(SMART).Diabetes Care,2007,30(6):1581 -1583.
23 Ruilope LM,Aldigier JC,Ponticelli C,et al.Safety of the combination of valsartan and benazepril in patients with chronic renal disease.European Group for the Investigation of Valsartan in Chronic Renal Disease.J Hypertens,2000,18(1):89 - 95.
