自噬与肾脏疾病
2012-05-10综述曾彩虹审校
范 芸 综述 曾彩虹 审校
近10年随着酵母模型和基因技术发展,人们对自噬分子机制和形态特点的研究逐渐深入。自噬是广泛存在于真核细胞的一种溶酶体依赖性蛋白降解过程:由孤立双层膜包裹部分胞质及需降解的细胞器和蛋白质形成自噬体,与细胞内溶酶体融合形成自噬性溶酶体,进而降解自噬体内膜结构及其内容物,实现细胞器更新;在清除蛋白质过程中产生游离氨基酸和脂肪酸再次供细胞利用,维持细胞内稳态。自噬作为一种细胞保护机制,其异常可导致多种疾病的发生、发展(如肿瘤、肾脏疾病等)。近年来,众多研究表明自噬在多种肾脏疾病进程中可能发挥着重要作用,但其变化和作用机制尚不明确。本文就自噬及其在肾脏疾病领域的研究进展作一综述。
自噬的分类
自噬主要包括微自噬、巨自噬和分子伴侣介导的自噬三种类型[1]。微自噬即溶酶体膜直接内陷将底物包裹并降解的过程。巨自噬即自噬体包裹需降解的底物,与溶酶体融合形成自噬性溶酶体,最后底物由溶酶体内水解酶降解的过程。分子伴侣介导的自噬即部分分子伴侣引导待降解蛋白转入溶酶体降解的过程。本文中所指的自噬主要是巨自噬(以下简称自噬)。
自噬的形成过程分四个阶段[2],(1)隔离膜形成:在各种自噬诱导因素刺激下,需降解的细胞器或蛋白质周围形成隔离膜;(2)自噬体形成:隔离膜逐渐延伸,将需降解的胞质成分完全包绕形成自噬体;(3)自噬性溶酶体形成:自噬体将其包裹物运输至溶酶体并与之融合形成自噬性溶酶体;(4)自噬体内容物降解:自噬体与溶酶体融合后,自噬体内膜及其内容物被溶酶体中多种蛋白水解酶降解(图1)。生理状态下几乎所有细胞都存在基础水平自噬现象,参与调控生物体的生长发育和细胞分化过程,维持细胞内环境稳定,降解蛋白质产生氨基酸等供细胞再循环,清除未折叠或折叠错误的蛋白质,从而保护细胞,甚至可以抗饥饿、抗衰老、抑制肿瘤和清除微生物等。
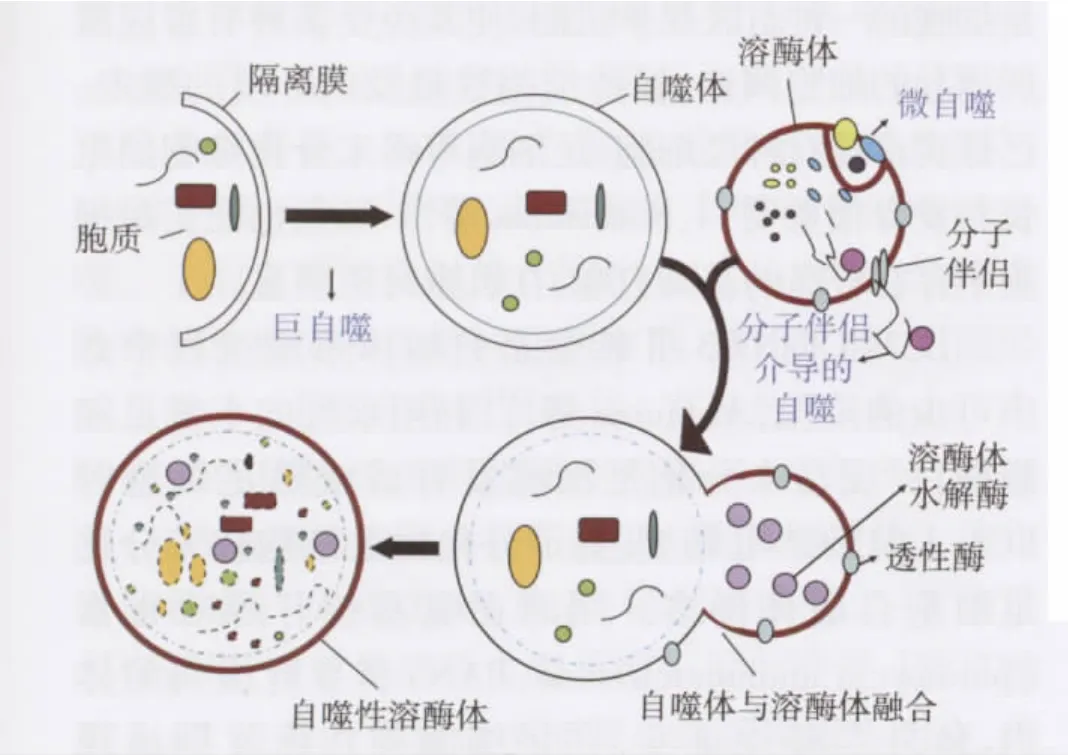
图1 自噬的形成过程[1,3]
信号调控
自噬是一种受严格调控的生理现象(图2)[4,5],在细胞正常生理活动和疾病过程中具有重要作用,了解其分子机制对于研究或探索自噬与部分疾病的相互关系意义重大。
哺乳类雷帕霉素靶蛋白(mTOR)信号通路
mTOR下游通路 mTOR激酶是自噬的主要调节剂,在营养充足如富含氨基酸和生长因子时,细胞中mTOR被激活,阻断自噬通路;而当营养缺乏,细胞处于饥饿环境时,mTOR活性受抑,解除对自噬通路的抑制。mTOR作为自噬抑制剂,激活后能抑制自噬相关基因或蛋白复合体的形成,包括Unc-51样激酸酶(ULK)/自噬基因(Atg1)复合体和Ⅲ型磷脂酰肌醇3激酶(PI3K-Ⅲ)蛋白复合体,两者都是自噬隔离膜形成的重要复合物。mTOR还可干扰促进自噬形成的两条关键通路——Atg8/微管相关蛋白1 轻链 3(LC3)通路和 Atg12-Atg5-Atgl6 通路。mTOR活化后还可调节两个下游因子-核糖体S6蛋白激酶1(S6K1)和真核翻译起始因子4E结合蛋白1(4EBP-1),两者是蛋白质翻译的关键调节因子,影响相关蛋白的转录和翻译来抑制自噬。
mTOR上游通路 (1)PI3K-Ⅰ/蛋白激酶 B(Akt/PKB)信号通路:胰岛素、生长因子结合跨膜胰岛素受体或酪氨酸激酶受体后,PI3K-Ⅰ/Akt信号被激活,抑制下游的结节性硬化复合物(TSC)来激活mTOR活性,发挥抑制自噬的作用。(2)AMP-依赖性蛋白激酶(AMPK)通路:AMPK是代谢性应激(包括葡萄糖缺乏,缺氧,局部缺血或氧化应激)下加强细胞自噬的重要因子。激活的AMPK通过直接磷酸化mTOR或激活TSC(TSC1/TSC2异源二聚体)抑制mTOR活性,从而达到增强自噬的目的。(3)氨基酸信号通路:氨基酸是自噬通路的主要终产物,部分氨基酸如丙氨酸、亮氨酸、谷氨酸盐和苯丙氨酸同时也是自噬的重要调节因子。氨基酸通过负性调控自噬水平,从而更好的借助自噬来维持细胞内环境稳定。
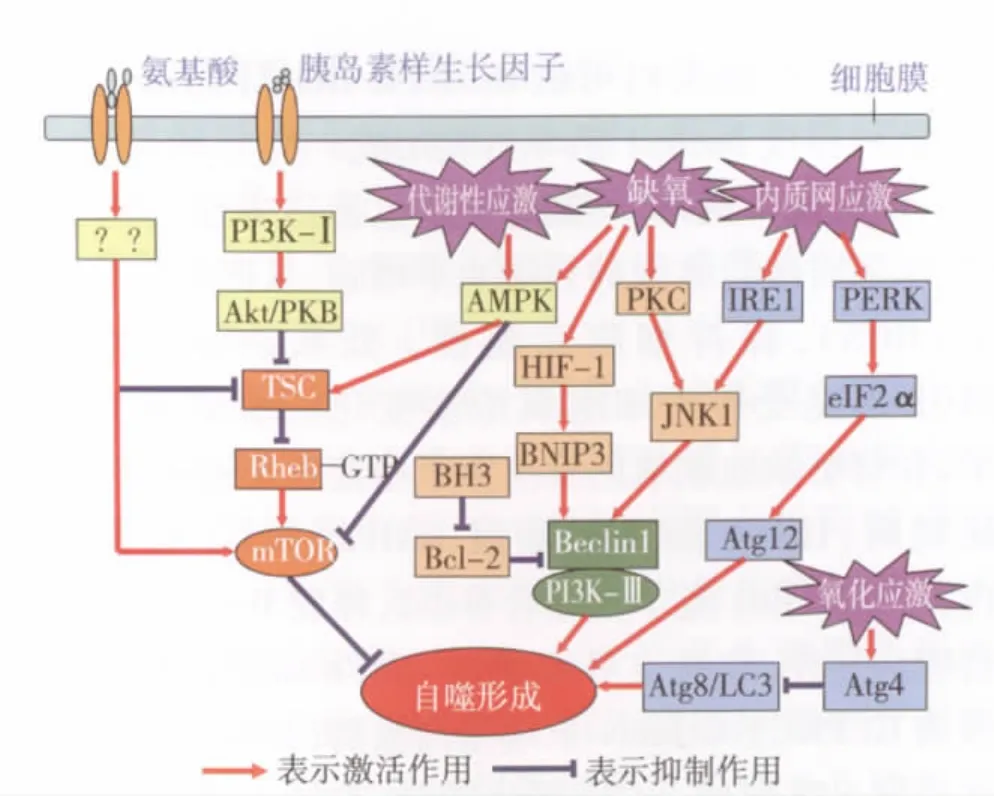
PI3K-Ⅰ:Ⅰ型磷脂酰肌醇3激酶;Akt/PKB:蛋白激酶B;TSC:结节性硬化复合物;AMPK:AMP-依赖性蛋白激酶;GTP:三磷酸鸟苷;Bcl-2:B细胞淋巴瘤/白血病基因2,Beclin1:酵母 Atg6同源物;PI3K-Ⅲ:Ⅲ型磷脂酰肌醇3激酶;HIF-1:低氧诱导因子1;IRE1:肌醇需酶1;PERK:PKR样ER调节激酶;PKC:蛋白激酶C-JNK1:c-jun氨基酸末端激酶;eIF2α:真核细胞翻译起始因子2α;BNIP3:腺病毒E1B19000相互作用蛋白3
B细胞淋巴瘤/白血病基因2(Bcl-2)-酵母Atg6同源物(Beclin1)通路 Beclin1是自噬过程中最重要的正调节因子,与PI3K-Ⅲ形成复合体调节其他Atg蛋白在自噬前体结构即隔离膜中的定位,参与自噬体的形成。研究表明Bcl-2家族蛋白是缺血缺氧等多种疾病状态下主要的非mTOR的自噬调节通路。Bcl-2是一种抗凋亡蛋白,定位于线粒体、内质网和核膜,在胞质内含量较少,Bcl-2结合到Beclin1的功能性结构域上抑制Beclin1与PI3K-Ⅲ相互作用,影响自噬形成。其同源染色体Bcl-XL、Bcl-w,Mcl-1 也对自噬具有抑制作用。而 Bcl-2 家族中的唯 BH3域蛋白可破坏 Bcl-2/Bcl-Xl与 beclin1的结合,释放beclin1诱导自噬形成。
细胞应激通路 细胞应激包括三方面:(1)缺氧:缺氧时线粒体内的自噬水平增强,从而降低活性氧(ROS),保持细胞完整性。低氧诱导因子1(HIF-1)是受控于细胞氧浓度变化的重要调节因子,在肾脏缺血缺氧的反应调节中起重要调控作用。缺氧时,HIF-1 激活靶基因 BNIP3(Bcl-2 家族蛋白),与beclin1竞争性结合Bcl-2,释放Beclin1激活自噬。另外,缺氧也可以通过AMPK-mTOR和蛋白激酶 C(PKC)-C-JunN-末端蛋白激酶(JNK1)信号通路诱导自噬形成。(2)氧化应激:ROS是细胞应激中一种常见的自噬诱导剂,包括超氧阴离子、自由基和过氧化物等。氧化应激时ROS可以抑制Atg4的蛋白酶活性,促进Atg8/LC3的脂化来激活自噬。(3)内质网应激:PERK[protein kinase(PKR)-like ER kinase]、IRE1(inositol requiring 1)是哺乳动物内质网应激时介导自噬形成的主要物质[6,7]:①IRE1-XBP1/IRE1-JNK途径:IRE1通路激活后释放其核酸内切酶的活性,导致XBP1 mRNA剪切,编码有活性的转录因子,上调多种基因编码Atg5、Atg7、Atg8和Atg19,促进自噬。IRE1还可募集肿瘤坏死因子受体相关因子2(TRAF-2)使下游的JNK磷酸化,激活JNK来诱导自噬形成。②内质网类似激酶(PERK)/真核细胞翻译启动子2α(eIF2α)途径:内质网应激时PERK磷酸化 eIF2α,上调 Atg12 形成 Atg5-Atg12-Atg16 复合物,引起LC3的膜转位诱发自噬[7]。
自噬与肾脏疾病
近年来,自噬在肿瘤及神经系统方面研究广泛,多种神经退行性疾病[8-10]和肿瘤[11]均与自噬异常有关,但其在肾脏疾病中的相关研究较少。1983年,Berkenstam等[12]首次从大鼠肾脏皮质中分离出自噬性囊泡。2003年,Asanuma等[13]发现自噬存在于足细胞的分化及损伤修复过程。2007年,Chien等[14]证实了自噬参与缺血-再灌注肾损伤的发生。近来自噬在肾脏疾病中的作用备受关注。
足细胞损伤及足细胞疾病 足细胞是肾小球滤过屏障的重要组成部分,是分裂和增生能力较差的终末分化细胞,毒物、免疫损伤、感染和氧化应激等均可使其足细胞损伤,使其功能障碍甚至凋亡[15]。足细胞数量减少,可作为预测疾病进展的指标之一[15,16]。足细胞损伤可导致多种疾病,临床治疗棘手,因此保护足细胞直接决定疾病转归。自噬激活是细胞的一种自我保护机制,使其免受多种有害应激所诱导的细胞凋亡,保持细胞数量及内环境的稳定。已证实自噬对神经细胞、足细胞等终末分化细胞的生长和发育很重要[3],Mizushima 等[17]研究也证实足细胞中存在较高的基础自噬,在饥饿时更明显。
LC3-Ⅰ向LC3-Ⅱ转变是自噬体形成过程中必不可少的阶段,Asanuma等[13]利用永生的小鼠足细胞株,发现在未分化足细胞发育成成熟足细胞时LC3-Ⅰ向LC3-Ⅱ转变,提示分化的足细胞较未分化足细胞自噬体增多。同时在氨基核苷嘌呤霉素(puromycin aminonucleoside,PAN)诱导肾损伤的体内、体外实验中证实,在足细胞损伤恢复期出现LC3-Ⅰ向LC3-Ⅱ的转变,推测自噬在足细胞的分化和损伤及恢复过程中起着重要的作用,但未进一步研究自噬与足细胞分化及PAN诱导的足细胞损伤之间的机制。
研究发现随着年龄增长,肾功能逐渐下降,肾组织结构发生相应改变,包括足细胞丢失和肾小球硬化[18],而衰老细胞或动物模型中能观察到自噬形成减少[19],目前并不清楚衰老足细胞中自噬减少是由细胞外还是细胞内通路介导。Hartleben等[20]特异性敲除足细胞自噬相关基因Atg5构建了足细胞自噬缺乏的小鼠模型,模拟衰老足细胞的表型。随着小鼠年龄增长,足细胞胞质内脂褐素增加、泛素化蛋白聚集、线粒体损伤及氧化蛋白负荷增加,从而表明自噬可清除蛋白聚集物对细胞产生保护作用。足细胞Atg5特异性敲除后,小鼠自噬功能减弱,使得高龄小鼠体内氧化蛋白蓄积,出现内质网应激和蛋白尿,最终导致足细胞损伤、丢失及肾小球硬化。与对照组相比,自噬缺乏的高龄小鼠局灶或球形硬化明显增多。该作者还对微小病变性肾病(MCD)、局灶节段性肾小球硬化(FSGS)和膜性肾病(MN)等患者进行观察,均发现肾组织中自噬增高。这些结果都提示自噬在一定程度上能够保护足细胞,自噬缺乏导致足细胞损伤及肾小球硬化加重。但也有研究报道自噬是Ⅱ型细胞程序性死亡程序,自噬的过度发生会导致细胞死亡,促进疾病的进展[4]。所以对足细胞疾病中自噬作用的深入研究,有助于将自噬作为治疗目标,从而改善肾小球疾病及年龄相关的肾功能减退。
IgA肾病(IgAN)IgAN是我国最常见肾小球疾病,占原发性肾小球肾炎的45.3%,占总体肾小球疾病的33.2%[21]。IgAN临床及病理表现不一,预后也存在差异,国外报道10年肾存活率为63%~96%[22],我国为 85%[23],因此如何评估 IgAN 的预后非常重要。Sato等[24]发现足细胞自噬类型与IgAN的预后关系密切,足细胞有Ⅰ型和Ⅱ型两类自噬。Ⅰ型自噬的自噬体(直径约1 μm)内包含较多致密的核糖体和少量的脂质空泡,膜不完整,不能形成自噬体。Ⅱ型自噬的自噬体(直径约3~8 μm)内致密的核糖体较少但脂质空泡较多,可完成自噬过程,有效地清除细胞内损伤的蛋白质及脂质体。该作者还观察了16例IgAN患者,每例均接受2~3次重复肾活检,重复肾活检自噬类型未发生改变,Ⅰ型自噬的IgAN患者大多数以中度肾小球系膜增生为主,伴轻~中度节段肾小球硬化,轻~中度肾小管萎缩,慢性化病变较Ⅱ型自噬为主的IgAN患者重,提示Ⅰ型自噬为主的IgAN患者疾病进展快,预后较差[25]。
糖尿病肾病(DN)DN是最常见继发性肾小球疾病之一,其发病率呈上升趋势,可进展为终末期肾病。研究表明足细胞超微结构改变及其分子表达变化在DN蛋白尿的发生发展中起着重要作用,已知自噬与足细胞内环境稳定有着密切联系,提示足细胞自噬在DN中可能发挥了重要作用。也有研究报道缺氧、内质网应激和氧化应激是DN的主要发病机制之一[26],而营养传感通路如 mTOR、AMPK、沉默信息调节因子同源物1(Sirt1)信号通路也参与了DN的发病[27],这些通路同样都是自噬形成的重要调控因素,说明自噬可能是DN的发病机制之一,但具体机制不清楚。Kitada等[27]发现2型糖尿病模型Wistar大鼠(WFR组)肾组织可见p62/SQSTM1聚集,近端肾小管细胞出现线粒体损伤。而p62/SQSTM1主要通过自噬性溶酶体降解,提示WFR组肾组织中自噬通路受抑制。WFR组还观察到Sirt1降低,Sirt1是自噬的正调节分子,这也从反面证实了WFR组自噬体形成减少。这些结果都表明DN存在自噬异常,但作用机制不明。Wu等[28]研究也发现自噬异常在早期DN发病中扮演了重要角色。
目前,DN中mTOR信号通路研究较多,mTOR信号通路是调节细胞体积的主要机制,其激活可能导致糖尿病患者足细胞肥大。研究表明糖尿病患者mTOR过度激活介导持续的信号刺激,导致足细胞功能退化、肾小球硬化及蛋白尿。mTOR为自噬的重要负性诱导剂,目前已经证实mTOR信号通路参与了多囊肾、肾肿瘤及遗传性肾小球疾病的发生,并与DN的蛋白尿及肾小球硬化的发病机制有关[29,30]。由此可推测 mTOR-自噬通路可能成为肾小球疾病的主要标记物之一。
自身免疫性肾脏病 抗肾小球基膜(GBM)肾炎,ANCA相关性血管炎及狼疮性肾炎等自身免疫性肾病的发病率不低,自噬(体内蛋白质降解的主要途径之一)可能为机体提供了自身免疫刺激分子和自身抗原的来源。Anders等[31]认为肾损伤后死亡细胞释放的蛋白质成分,如危险信号相关分子模式(DAMPs),作为免疫刺激因子可识别Toll样受体(TLRs)和其他的固有免疫受体,促使细胞因子和趋化因子的分泌,白细胞募集及组织重塑等。在肾小管上皮细胞或树突状细胞和巨噬细胞产生的DAMPs和病原相关分子模式(PAMPs)主要通过自噬途径进行降解。在自噬性溶酶体中,DAMPs和PAMPs可被细胞表面的TLRs和其他固有免疫受体识别,然后通过一系列蛋白质级联反应激活转录因子核因子 κB(NF-κB)和 Jun/Fos,从而有效地激活免疫反应。若自噬性溶酶体内的DAMPs或PAMPs是体内的隐蔽抗原,通过识别TLRs和主要组织相容性复合物(MHC-Ⅰ或MHC-Ⅱ)成为自身抗原,就能导致免疫耐受缺失和自身免疫性疾病。
中毒性肾损伤 局部缺血、毒素、脓毒症引起的急性肾损伤是典型的肾脏应激表现,可导致肾脏细胞死亡、组织损伤及肾功能下降。Chien等[14]和Suzuki等[32]研究发现小鼠缺血再灌注肾损伤和人移植肾组织中自噬相关基因表达升高,证实存在自噬异常,但起保护作用还是致病作用存在争议。顺铂是常用的化疗药物之一,具有明显肾毒性,可导致肾小管上皮细胞凋亡、坏死和急性肾损伤。Periyasamy-Thandavan 等[33]利用经顺铂处理培养的人近端肾小管(RPTC)建立细胞应激、损伤、死亡模型,在顺铂处理早期观察到RPTC自噬的激活、自噬体增多,早于细胞凋亡及肾脏损伤:细胞经顺铂处理数小时后即可以出现自噬,而在处理后12h内未检出细胞凋亡,说明顺铂介导的自噬非凋亡所致或继发于凋亡。抑制自噬显著增加顺铂诱导的细胞凋亡,提示自噬在顺铂诱导的肾小管上皮细胞早期损伤中发挥保护作用。在同一研究中,随着顺铂处理时间延长,C57BL/6小鼠肾组织中自噬性囊泡也在增加,进一步在体内证实了自噬对RTPC的保护作用。
环孢素A(CsA)广泛用于预防器官移植后的急性排斥及治疗自身免疫性疾病,但是长期使用可导致慢性肾脏损伤,包括间质纤维化,肾小管萎缩及肾小球硬化等病变。Pallet等[34]证实CsA通过内质网应激激活肾小管上皮细胞的自噬活性,在CsA处理早期自噬激活可起到保护肾小管上皮细胞的作用,减轻CsA对肾小管上皮细胞的毒性作用。以上研究都提示自噬对肾小管上皮细胞具有保护作用,为寻找保护肾小管上皮细胞的研究提供了基础。
展 望
自噬是贯穿于细胞生长发育和生理病理活动的重要过程,随着对自噬研究的不断深入,其在肾脏疾病发生、发展中的作用日渐引起关注,但自噬以何种机制来引起或改善肾脏疾病尚不明了,因此对肾脏病中自噬作用机制的研究具有重要的理论意义和应用价值。已知敲除自噬相关基因,可增加细胞对放疗的敏感度,为肿瘤治疗提供新靶点。我们期望通过对自噬在肾脏疾病发生中的作用研究,为难治性肾脏疾病找到新临床治疗途径。
1 Mizushima N,Yoshimori T,LevineB.Methods inmammalian autophagy research.Cell,2010,140(3):313 - 326.
2 Reggiori F,Klionsky DJ.Autophagy in the eukaryotic cell.Eukaryot Cell,2002,1(1):11 -21.
3 Mizushima N,Levine B,Cuervo AM,et al.Autophagy fights disease through cellular self-digestion.Nature,2008,451(7182):1069 -1075.
4 Periyasamy-Thandavan S,Jiang M,Schoenlein P,et al.Autophagy:molecular machinery, regulation, and implications for renal pathophysiology.Am J Physiol Renal Physiol,2009,297(2):F244 -256.
5 He C,Klionsky DJ.Regulation mechanisms and signaling pathways of autophagy.Annu Rev Genet 2009,43:67 -93.
6 Kouroku Y,Fujita E,Tanida I,et al.ER stress(PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion,an essential step for autophagy formation.Cell Death Differ,2007,14(2):230 -239.
7 Ogata M,Hino S,Saito A,et al.Autophagy is activated for cell survival after endoplasmic reticulum stress.Mol Cell Biol,2006,26(24):9220-9231.
8 Yang DS,Kumar A,Stavrides P,et al.Neuronal apoptosis and autophagy cross talk in aging PS/APP mice,a model of Alzheimer's disease.Am J Pathol,2008,173(3):665 -681.
9 Pickford F,Masliah E,Britschgi M,et al.The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice.J Clin Invest,2008,118(6):2190-2199.
10 Tannous P,Zhu H,Nemchenko A,et al.Intracellular protein aggregation isa proximaltriggerofcardiomyocyte autophagy.Circulation,2008,117(24):3070 -3078.
11 Levine B.Cell biology:autophagy and cancer.Nature,2007,446(7137):745-747.
12 Berkenstam A,Ahlberg J,Glaumann H.Isolation and characterization of autophagic vacuoles from rat kidney cortex.Virchows Arch B Cell Pathol Incl Mol Pathol,1983,44(3):275 - 286.
13 Asanuma K,Tanida I,Shirato I,et al.MAP-LC3,a promising autophagosomal marker,is processed during the differentiation and recovery of podocytes from PAN nephrosis.FASEB J,2003,17(9):1165-1167.
14 Chien CT,Shyue SK,Lai MK.Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy.Transplantation,2007,84(9):1183 -1190.
15 Vogelmann SU,Nelson WJ,Myers BD,et al.Urinary excretion of viable podocytes in health and renal disease.Am J Physiol Renal Physiol,2003,285(1):F40 -48.
16 White KE,Bilous RW,Marshall SM,et al.Podocyte number in normotensive type 1 diabetic patients with albuminuria.Diabetes,2002,51(10):3083 -3089.
17 Mizushima N,Yamamoto A,Matsui M,et al.In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker.Mol Biol Cell,2004,15(3):1101-1111.
18 Wiggins JE,Goyal M,Sanden SK,et al.Podocyte hypertrophy,“adaptation,”and“decompensation”associated with glomerular enlargement and glomerulosclerosis in the aging rat:prevention by calorie restriction.J Am Soc Nephrol,2005,16(10):2953 -2966.
19 Cuervo AM,Bergamini E,Brunk UT,et al.Autophagy and aging:the importance ofmaintaining“clean” cells.Autophagy,2005,1(3):131-140.
20 Hartleben B,Gödel M,Meyer-Schwesinger C,et al.Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice.J Clin Invest,2010,120(4):1084 - 1096.
21 Li LS,Liu ZH.Epidemiologic data of renal diseases from a single unit in China:analysis based on 13,519 renal biopsies.Kidney Int,2004,66(3):920-923.
22 Geddes CC,Rauta V,Gronhagen-Riska C,et al.A tricontinental view of IgA nephropathy.Nephrol Dial Transplant,2003,18(8):1541 -1548.
23 Le W,Liang S,Hu Y,et al.Long-term renal survival and related risk factors in patients with IgA nephropathy:results from a cohort of 1155 cases in a Chinese adult population.Nephrol Dial Transplant,2012,27(4):1479-1485.
24 Sato S,Yanagihara T,Ghazizadeh M,et al.Correlation of autophagy type in podocytes with histopathological diagnosis of IgA nephropathy.Pathobiology,2009,76(5):221 -226.
25 Sato S,Kitamura H,Adachi A,et al.Two types of autophagy in the podocytesin renalbiopsy specimens:ultrastructuralstudy.J Submicrosc Cytol Pathol,2006,38(2 -3):167 -174.
26 Wu J,Zhang R,Torreggiani M,et al.Induction of diabetes in aged C57B6 mice results in severe nephropathy:an association with oxidative stress,endoplasmic reticulum stress,and inflammation.Am J Pathol,2010,176(5):2163 -2176.
27 Kitada M,Takeda A,Nagai T,et al.Dietary Restriction Ameliorates Diabetic Nephropathy through Anti-Inflammatory Effects and Regulation of the Autophagy via Restoration of Sirt1 in Diabetic Wistar Fatty(fa/fa)Rats:A Model of Type 2 Diabetes.Exp Diabetes Res,2011,2011:908185.
28 Wu WH,Zhang MP,Zhang F,et al.The role of programmed cell death in streptozotocin-induced early diabetic nephropathy.J Endocrinol Invest,2011,34(9):e296 - 301.
29 Gödel M,Hartleben B,Herbach N,et al.Role of mTOR in podocyte function and diabetic nephropathy in humans and mice.J Clin Invest,2011,121(6):2197 -2209.
30 Inoki K,Mori H,Wang J,et al.mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice.J Clin Invest,2011,121(6):2181 -2196.
31 Anders HJ,Schlondorff DO.Innate immune receptors and autophagy:implications for autoimmune kidney injury.Kidney Int,2010,78(1):29-37.
32 Suzuki C,Isaka Y,Takabatake Y,et al.Participation of autophagy in renal ischemia/reperfusion injury.Biochem Biophys Res Commun,2008,368(1):100 -106.
33 Periyasamy-Thandavan S,Jiang M,Wei Q,et al.Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells.Kidney Int,2008,74(5):631 -640.
34 Pallet N,Bouvier N,Legendre C,et al.Autophagy protects renal tubular cells against cyclosporine toxicity.Autophagy,2008,4(6):783-791.
