抗菌药物新靶位
2012-04-13陈盈盈张玉彬顾觉奋
陈盈盈,张玉彬,顾觉奋
·综述·
抗菌药物新靶位
陈盈盈,张玉彬,顾觉奋
近年来,由于过度使用抗生素,特别是滥用抗生素,病原菌日益广泛的耐药性已成为药物治疗中越来越常见的问题[1]。为了克服细菌的耐药性,特别是多重耐药性问题,人们需要运用新的思路去发掘新的抗生素,因此,探索新型抗菌机制已成为目前的迫切需要。本文综述了近几年发现的抗菌药物的新靶位,分别从作用于细菌(真菌)细胞壁、细胞膜、RNA 聚合酶、胞内代谢及调控等 4 方面来谈抗菌药物的新作用靶位。
1 细胞壁
肽聚糖(peptidoglycan)是构成细菌细胞壁的主要成分,革兰阳性和阴性细菌中都含有肽聚糖结构,因此肽聚糖的生物合成途径成为开发广谱抗菌药物的研究热点。
D-丙氨酸是细菌细胞壁中肽聚糖结构的组成成分,主要存在于二肽 D-丙氨酰-D-丙氨酸中,D-丙氨酸直接参与相邻肽聚糖链的交联。在原核生物中,D-丙氨酸的主要合成途径是通过丙氨酸消旋酶(Alr)催化 L-丙氨酸转化而来[2]。
催化肽聚糖合成的胞浆步骤的酶包括烯醇丙酮转移酶MurA、黄素依赖还原酶 MurB 和 ATP 依赖的连接酶MurC、MurD、MurE 和 MurF,通过这些酶的作用,为UDPMurNAc 乙酰基侧链添加 L-丙氨酸、D-谷氨酸、内消旋二氨基庚二酸(M-DAP)或 L-赖氨酸、D-丙氨酸-D-丙氨酸二肽,生成 UDPMurNAc-五肽,之后移位酶 MraY 和糖基转移酶 MurG 催化 UDPMurNAc-五肽转化为脂质中间体 I 和 II。
某些革兰阳性菌酶,如金黄色葡萄球菌 FemABX 酶以甘氨酰-tRNA 为底物,增加 5个甘氨酸残基,参与肽交联;肺炎链球菌 MurM 和 MurN 连接酶分别向脂质中间体 II添加 Ala/Ser 和 Ala。虽然这些酶不是广谱抗菌的靶位,但这些酶的抑制剂可能用于与青霉素协同治疗耐青霉素病原体。
李冬和李卓荣[3]报道,5-氨基-呋喃糖苷衍生物大多有抗分枝杆菌的活性,也可以有效抑制丙氨酸消旋酶,其最低抑菌浓度为 3112 μg/ml。Sova 等[4]发现 1-(4-甲氧苯基磺酰氨基-)3-吗啉丙烷-2-磷酸二氢(图 1),是目前为止最好的MurE 连接酶抑制剂,其 IC50为 6 μmol/L。其他作用于肽聚糖生物合成途径的靶点及其抑制剂如表 1 所示。
2 细胞膜
细菌中 FASII 系统的单功能酶已成为基因组驱动的新型抗菌药物靶标研究的热点[5]。以大肠杆菌为例,细菌脂肪酸生物合成(fatty acid biosynthesis,Fab)的途径如图 2 所示。FabH 和 FabI 与相应的人类酶相比,缺乏总的序列的同源性,因此成为抗菌药物极好的靶位[6]。

图1 1-(4-甲氧苯基磺酰氨基)-3-吗啉丙烷-2-磷酸二氢

表1 肽聚糖生物合成酶抑制剂的效力(IC50)和抗菌活性(MIC)
FabH 介导乙酰辅酶 A 和丙二酰 ACP 缩合形成β-酮酰基-ACP,是催化脂肪酸合成碳链延长循环的起始因子,这一调控步骤在确定 FASII 途径产生的脂肪酸量也起到了关键作用。FabI 是细菌脂肪酸生物合成中必需的酶,其催化反式-2-烯酰-ACP 还原为酰基-ACP,为延伸循环的最后步骤,也是限速步骤。
张学辉等[7]报道了关于 FabH 的相关抑制剂。Wen 等[8]从 46 种新合成的 1,6 二取代吲哚 2-羧酸类似物中筛选得到 WIUAKP-001,对 FT-FabH 酶抑制的 IC50为 2 μmol/L,WIUAKP-031 和 WIUAKP-056 对 FT-FabI 酶抑制活性的IC50分别为 6 μmol/L 和 7 μmol/L(表 2)。
3 RNA 聚合酶
细菌的 RNA 聚合酶(RNAP)内转化区是一个新发现的药物靶标,作用于此靶位的酶抑制剂不存在和其他抗菌药物交叉耐药的现象。
转换区是一个结构元件,在转录起始阶段,DNA 加载到 RNA 聚合酶活跃的中心裂缝时,介导 RNA 聚合酶构象的变化[9-11](图 3)。转化区位于 RNA 聚合酶钳夹的底部,是铰链调节 RNA 聚合酶的开关。当 DNA 要加载到RNA 聚合酶活跃的中心裂缝时,钳夹打开,当 DNA 停留在 RNA 聚合酶裂缝中心时,钳夹关闭。转换区共有 5 部分,分别被称为“转换 1”到“转换 5”,它们局部构象的改变可以调控钳夹的开启和闭合(图 3B)[12]。在 RNA 聚合酶裂缝中心,转换 1、2、3 可以直接接触已展开的 DNA模板链[13]。
4 个和转换区结合的天然产物已被确定,均具有抑制细菌 RNA 聚合酶活性,并表现出广谱抗菌活性,它们分别是:myxopyronin(Myx)、corallopyronin(Cor)、ripostatin(Rip)和 lipiarmycin(Lpm)(表 3)[14]。根据 Myx/Cor/Rip 作用靶位,目前已筛选出 3 种新型的结构不相关的 RNA 聚合酶抑制剂。
4 胞内调控作用
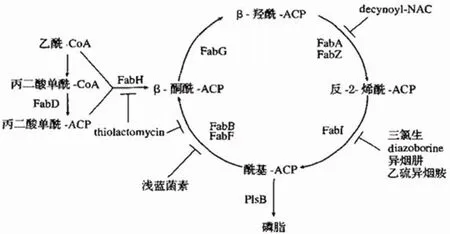
图2 大肠杆菌脂肪酸的生物合成途径

表2 酶抑制剂 WIUAKP-001、WIUAKP-031 和 WIUAKP-056 的结构和作用靶位
大多原核生物采用一种类似的机制—“双组分系统”调控蛋白质磷酸化,进行信号转导。迄今为止,此种机制尚未在高等真核生物体内发现。“双组分系统”的核心通常由两个蛋白组成。一是组氨酸蛋白激酶(HK)。通常它是一个跨膜蛋白,结构可分为 3 部分,自 N 端到 C 端依次为配体(信号)结合部位,为高度可变区;组氨酸自身磷酸化位点,分子构象为二聚体;ATP 结合的激酶功能区,内含 N、G1、F、G2 等 4 个保守的基序(motif)。另一个蛋白为反应调节蛋白(RR),是一个胞质蛋白。结构上分为两部分,N 端含有能接受磷酸基团的天冬氨酸位点,相对保守,为调控区域;C 端为效应区,序列高度可变,含有多个可与不同的 DNA 序列特异性结合的位点。
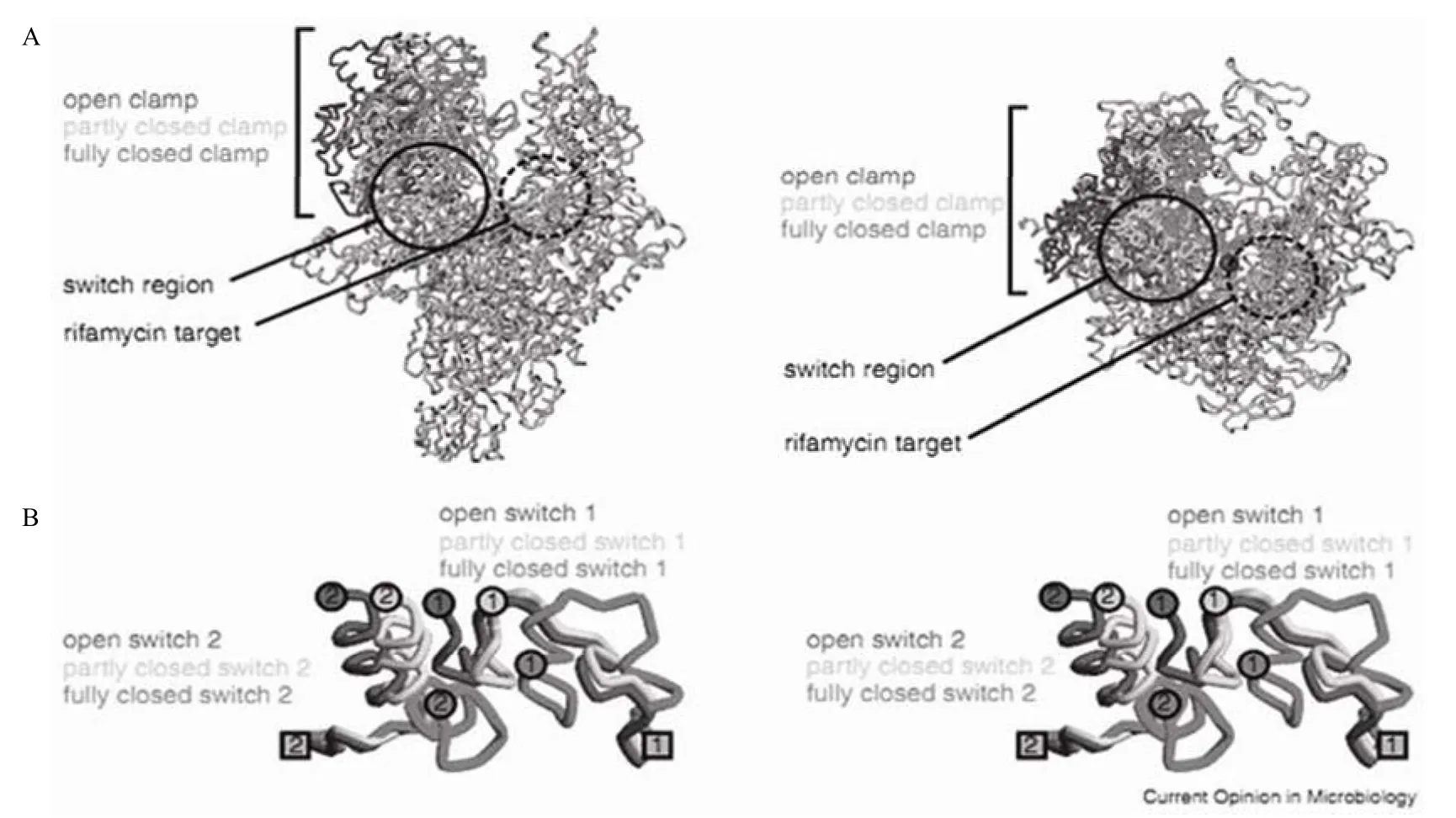
图3 RNA 聚合酶夹子和 RNA 聚合酶转化区[A:RNA 聚合酶钳夹的构象状态(两个正交视图)[12]。RNA 聚合酶结构呈现出开放(黑色),半封闭(浅灰色),并完全封闭(灰色)构象。实线圈代表转换区,虚线圈为利福霉素的结合位点,实线圈与虚线圈之间的黑点为 Mg2+的活性中心;B:RNA 聚合酶转化区构象的立体视图。RNA 聚合酶转换 1 和转换 2 的结构开放(黑色),半封闭(浅灰色),全封闭(灰色)钳夹构象。灰色方块为转换 1 和转换 2 与 RNA 聚合酶主体的连接点。黑、浅灰、灰色圆圈分别表示转换 1 和转换 2 与 RNA 聚合酶钳夹的连接点]

表3 作用在 RNA 聚合酶转换区的 RNA 聚合酶抑制剂的活性

图4 几种蛋白激酶抑制剂的化学结构
当外界信号作用于组氨酸蛋白激酶的膜外配体结合区时,激活了激酶的 ATP 结合部位、水解 ATP 为 ADP,并将 ATP 的磷酸基团转移到激酶上的组氨酸位点,使其发生自身磷酸化。随后,反应调节蛋白的调控区域能与磷酸化的组氨酸蛋白激酶相互作用,将激酶上的磷酸基团转移到自身的天冬氨酸位点上,发生自身磷酸化,并能激活效应区,使其构象改变,暴露不同的 DNA 结合位点,结合不同的DNA 序列,产生一系列的调控反应。针对作用在组氨酸激酶的作用靶点,归纳近几年其有效的抑制剂如图 4 和表 4所示。

表4 几种蛋白激酶的作用机制及其抑制剂
5 结论
细菌的耐药性及交叉耐药性日益严重,寻找新型作用机制的抗菌药物,发现新作用靶点及其有效的抑制剂是今后抗生素药物发展的一个重要方向。如上介绍了 4 种类型的作用靶位及其靶酶抑制剂,这些潜在药物目前大多仍处于对先导物优化的起始研究阶段,但人们对这些针对细菌耐药性的新思路及其发展前景充满信心。通过研究抗菌药物新靶位,推出新的高效抗菌药物,将为人类重大疾病提供突破性治疗。
参考文献
[1] Alekshun MN, Levy SB. Molecular mechanisms of antibacterial multidrug resistance. Cell, 2007, 128(6):1037-1050.
[2] Chacon O, Bermudez LE, Zinniel DK, et al. Impairment of D-alanine biosynthesis in Mycobacterium smegmatis determines decreased intracellular survival in human macrophages. Microbiology, 2009, 155(Pt 5):1440-1450.
[3] Li D, Li ZR. Progress in the research of antituberculous drugs (I). Chin J New Drugs, 2009, 18(1):35-42. (in Chinese)李冬, 李卓荣. 抗结核药物的研究进展(一). 中国新药杂志, 2009, 18(1):35-42.
[4] Sova M, Kovac A, Turk S, et al. Phosphorylated hydroxyethylamines as novel inhibitors of the bacterial cell wall biosynthesis enzymes MurC to MurF. Bioorg Chem, 2009, 37(6):217-222.
[5] Zhang YM, White SW, Rock CO. Inhibiting bacterial fatty acid synthesis. J Biol Chem, 2006, 281(26):17541-17544.
[6] Lu H, Tonge PJ. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc Chem Res, 2008, 41(1):11-20.
[7] Zhang XH, Yu H, Wang LL, et al. Progress in the research of antibacterial drugs new target FabH and its inhibitors. Foreign Med Sci (Section Pharm), 2006, 33(4):262-265. (in Chinese)张学辉, 于红, 王莉莉, 等. 抗菌药物新靶标FabH及其抑制剂研究进展. 国外医学药学分册, 2006, 33(4):262-265.
[8] Wen L, Chmielowski JN, Bohn KC, et al. Functional expression of Francisella tularensis FabH and FabI, potential antibacterial targets. Protein Expr Purifi, 2009, 65(1):83-91.
[9] Belogurov GA, Vassylyeva MN, Sevostyanova A, et al. Transcription inactivation through local refolding of the RNA polymerase structure. Nature, 2009, 457(7227):332-335.
[10] Tupin A, Gualtieri M, Brodolin K, et al. Myxopyronin: a punch in the jaws of bacterial RNA polymerase. Future Microbiol, 2009, 4(2): 145-149.
[11] Haebich D, von Nussbaum F. Lost in transcription--inhibition of RNA polymerase. Angew Chem Int Ed Engl, 2009, 48(19):3397-3400.
[12] Mukhopadhyay J, Das K, Ismail S, et al. The RNA polymerase “switch region” is a target for inhibitors. Cell, 2008, 135(2):295-307.
[13] Vassylyev DG, Vassylyeva MN, Perederina A, et al. Structural basis for transcription elongation by bacterial RNA polymerase. Nature, 2007, 448(7150):157-162.
[14] Tupin A, Gualtieri M, Leonetti JP, et al. The transcription inhibitor lipiarmycin blocks DNA fitting into the RNA polymerase catalytic site. EMBO J, 2010, 29(15):2527-2537.
[15] Gilmour R, Foster JE, Sheng Q, et al. New class of competitive inhibitor of bacterial histidine kinases. J Bacteriol, 2005, 187(23): 8196-8200.
[16] Qin Z, Zhang J, Xu B, et al. Structure-based discovery of inhibitors of the YycG histidine kinase: new chemical leads to combat Staphylococcus epidermidis infections. BMC Microbiol, 2006, 6:96.
[17] Kitayama T, Iwabuchi R, Minagawa S, et al. Synthesis of a novel inhibitor against MRSA and VRE: preparation from zerumbone ring opening material showing histidine-kinase inhibition. Bioorg Med Chem Lett, 2007, 17(4):1098-1101.
[18] Wehenkel A, Fernandez P, Bellinzoni M, et al. The structure of PknB in complex with mitoxantrone, an ATP-competitive inhibitor, suggests amode of protein kinase regulation inmycobacteria. FEBS Lett, 2006, 580(13):3018-3022.
[19] Székely R, Wáczek F, Szabadkai I, et al. A novel drug discovery concept for tuberculosis: inhibition of bacterial and host cell signaling. Immunol Lett, 2008, 116(2):225-231.
[20] Oxoby M, Moreau F, Durant L, et al. Towards Gram-positive antivirulence drugs: new inhibitors of Streptococcus agalactiae Stk1. Bioorg Med Chem Lett, 2010, 20(12):3486-3490.
DOI:10.3969/cmba.j.issn.1673-713X.2012.04.011
作者单位:210009 南京,中国药科大学生命科学与技术学院
通讯作者:顾觉奋,Email:yqyan1@126.com
收稿日期:2012-03-05
