噬菌体基因Ⅴ蛋白基因的合成、重组表达及功能分析
2012-04-01张大川房国梁陈江源刘烈炬刘志国
张大川,房国梁,李 琦,陈江源,王 岚,李 睿,刘烈炬,刘志国
(武汉工业学院生物与制药工程学院, 湖北 武汉 430023)
噬菌体基因Ⅴ蛋白基因的合成、重组表达及功能分析
张大川,房国梁,李 琦,陈江源,王 岚,李 睿,刘烈炬,刘志国*
(武汉工业学院生物与制药工程学院, 湖北 武汉 430023)
目的:研究丝状噬菌体基因V蛋白(gene V protein,GVP)基因的合成、重组表达及其功能。方法:根据GVP的基因序列,选择大肠杆菌偏爱的密码子,设计合成了8个寡核苷酸片段,利用重叠延伸PCR合成GVP基因序列,将其与原核表达载体pET-28a-c(+)质粒重组,转化大肠杆菌,获得GVP蛋白阳性表达菌株,异丙基-β-D-硫代吡喃半乳糖苷(IPTG)诱导表达,产物经Ni+-NTA琼脂糖凝胶层析纯化,获得目的蛋白GVP,DNA结合实验检测其功能。结果:成功合成出GVP基因,重组体在大肠杆菌BL21(DE3)中诱导获得高效表达,DNA结合实验表明GVP与单链DNA间解离平衡常数Kd=7.27×10-5mol/L。结论:重组构建并高效表达的GVP蛋白具有较高单链DNA结合能力,可用于食品病原微生物特定单链DNA分子的浓缩和分离。
基因V蛋白(GVP);大肠杆菌BL21(DE3);重组;表达
丝状噬菌体多可感染含F因子的大肠杆菌,基因V蛋白(gene V protein,GVP)主要是由Ff组的M13、F1、fd等丝状噬菌体产生的一种非结构蛋白,由87个氨基酸残基组成,为噬菌体DNA复制所必需,可使噬菌体DNA移动至细菌的细胞膜,属于单链DNA结合蛋白,而且结合单链核苷酸与碱基序列无直接关系,该蛋白还可抑制翻译活性,在病毒、原核生物的DNA复制、翻译及遗传重组等过程中发挥重要作用[1-3]。
已有研究表明,利用GVP与单链DNA非特异性结合的特性,在核酸与蛋白质相互作用、病原微生物的核酸检测以及DNA测序等许多方面可有广泛应用,如利用某些单链DNA结合蛋白筛选生物标本或样品中单链核苷酸片段来研究某些外显子的特性等[4-6]。可见该类DNA结合蛋白作为应用工具有较高的价值。
野生型的GVP广泛存在于细菌中,但含量较低且不易收集。本研究旨在利用重叠延伸PCR方法合成GVP基因序列,与质粒重组并在大肠杆菌中高效表达,产物经分离纯化后进行单链DNA(single strain DNA,ssDNA)结合力检测,为食品等病原微生物中单链特性的DNA片段分离、浓缩及检测奠定基础。
1 材料与方法
1.1 材料
大肠杆菌DH5α菌株、BL21(DE3)菌株、质粒pET-28a-c(+)由武汉工业学院生物技术研究室保存;限制性内切酶Xho I、EcoR I、T4连接酶 日本Toyobo公司;50bp DNA Ladder Marker 宝生物工程(大连)有限公司;1kb plus DNA Ladder 天根生化科技(北京)有限公司;PCR产物纯化试剂盒 美国Axygen公司;DNA凝胶回收试剂盒 上海捷瑞生物工程有限公司;Pfu DNA聚合酶、质粒小量制备试剂盒、中分子质量标准蛋白上海捷瑞生物工程有限公司;蛋白胨、酵母浸提物 英国Oxiod公司;Ni+-NTA树脂 Novagen公司;DNA引物由上海生工生物工程技术服务有限公司合成。
1.2 重组子pET-28a-c(+)-GⅤP的构建
利用重叠延伸PCR方法[7]构建目的基因,参照GeneBank上的GVP基因序列(Accession No. J02450),将目的基因中的大肠杆菌稀有密码子改为大肠杆菌偏爱密码子,在其5'端加EcoR I酶切位点,3'端加XhoI的酶切位点,两端各加两个保护碱基。整个基因序列全长280bp,将其分8段合成DNA引物,相邻两段间各有18个碱基互补重叠。
8段引物分别为:Pr1:5'- CGGAATTCATGATTAA AGTTGAAATT-3'下划线碱基为EcoRI酶切位点;Pr2:5'- GAAACACCAGAACGGGTGGTGAACTGCGCCTGA GACGGTTTAATTTCAACTTTAATCAT-3';Pr3:5'-CACCCGTTCTGGTGTTTCTCGTCAGGGCAAACCGT ATTCAGTGAATGAGCAGCTGTGTT-3';Pr4:5'-AATCTTGACCAGCACCGGATACTGATTACCCAGATCACGT AACACAGCTGCTCATTCA-3';Pr5:5'- CACCC GTTCTGGTGTTTCTCGTCAGGGCAAACCGTATTCAGT GAATGAGCAGCTGTGTT-3';Pr6:5'-AACC GAACTGACCAACTTTGAACGAGGACAGATGA ACGGTGTACAGACCCGGCGCATAG-3';Pr7:5'-AAGTTGGTCAGTTCGGTTCCCTGATGATTGATC GTCTGCGCCTGGTTCCGGCGAAATAA-3';Pr8:5'-ATCTCGAGTTATTTCGCCGGAACCAG-3'。下划线碱基为Xho I酶切位点。
利用相邻引物间重叠互补的核苷酸片段互相搭桥,并各为模板,进行3轮重叠延伸PCR[7],最终将8片段连成目的基因[8-9],以此目的基因为模板,Pr1和Pr8分别为上下游引物进行PCR,1%琼脂糖凝胶电泳检测扩增产物。
分别用EcoR I和Xho I于37℃水浴双酶切4h目的基因及质粒pET-28a-c(+),各自回收后,将酶切回收的目的基因和质粒用T4 DNA连接酶于16℃进行连接反应10h;连接产物即重组子转化大肠杆菌DH5α感受态细胞,涂布于含有卡那霉素(50μg/mL)的LB固体培养基上,筛选阳性菌落,提取质粒酶切并PCR扩增鉴定重组基因,并送交上海杰瑞生物工程有限公司测序检测。测序结果与设计序列相符的重组子命名为pET-28a-c(+)-GVP。
1.3 重组子pET-28a-c(+)-GVP的表达
将重组子pET-28a-c(+)-GVP转化大肠杆菌BL21(DE3)感受态细胞,涂布于含有卡那霉素(50μg/mL)的LB固体培养基上,挑取单菌落接种于含有卡那霉素(50μg/mL)的2mL LB液体培养基中37℃培养过夜,吸取少量过夜培养液按1:100的比例转接到新鲜LB(含有卡那霉素50μg/mL)培养基中,于37℃摇床培养至OD600nm为0.6~0.8,加入异丙基-β-D-硫代吡喃半乳糖苷(IPTG)(终浓度1.0mmol/L)诱导10h[10]。收集菌体提取蛋白进行SDSPAGE电泳检测。
1.4 重组蛋白的纯化
将诱导后的菌液于4℃、8000r/min离心20min,弃上清液,细菌沉淀用1/10的裂解液 (NaH2PO420mmol/L、NaCl 0.5mol/L、1% TritonX-100,pH7.4)重悬,超声波破碎,4℃、12000r/min离心20min,收集上清液,加入终质量浓度为5μg/mL的DNase和RNase,30℃水浴30min。使用Ni+-NTA琼脂糖层析柱纯化上清液中的目的蛋白,进行SDS-PAGE分析。
1.5 优化重组蛋白的表达条件
1.5.1 IPTG浓度对GVP蛋白表达的影响
按1.3节方法分别用浓度为0.2、0.4、0.6、0.8、1.0、1.2mmol/L的IPTG进行诱导4h,收集菌体蛋白进行SDS-PAGE电泳检测。
1.5.2 诱导时间对GVP蛋白表达的影响
根据1.5.1节所得最佳浓度的IPTG分别诱导1、2、4、6、8h,收集菌体蛋白进行SDS-PAGE电泳分析。
1.6 重组蛋白与单链DNA结合力的分析
纯化后的GVP蛋白与Ni+-NTA琼脂糖凝胶相偶联。偶联后的凝胶各取50μL,分别加入浓度分别为0.018、0.036、0.072、0.108、0.144、0.180mol/L 的pET-28a-c(+)单链DNA(质粒pET-28a-c(+)95℃变性10min,立即冰浴30min),混匀,20℃孵育1h,3000×g离心5min,弃上清液;用洗涤液(磷酸钠20mmol/L、NaCl 0.5mol/L,pH7.4)洗两次,加入100μL洗脱液(磷酸钠20mmol/L、NaCl 0.5mol/L、咪唑500mmol/L,pH7.4)洗脱,3000×g离心5min,取上清液。测上清液单链DNA浓度,绘制饱和曲线和Scatchard图。
2 结果与分析
2.1 目的基因的合成及重组基因的鉴定
经过第1轮重叠延伸PCR将8段引物合成4段核苷酸序列(图1)。纯化回收合成的4段序列,进行第2轮重叠延伸PCR,进一步合成两个更大的片段,最后第3轮重叠延伸PCR合成目的基因片段(图2)。提取转化的大肠杆菌DH5 α阳性菌落质粒,经双酶切和PCR扩增,阳性重组质粒可以切出与目的基因大小一致的片段,均为280bp(图3)。阳性重组质粒测序结果与设计完全相符,序列完整且阅读框正确。

图1 第1轮重叠延伸PCRFig.1 The first step of overlap extension PCR
2.2 重组质粒在大肠杆菌BL21(DE3)中的表达及重组蛋白的纯化
SDS-PAGE实验结果显示在相对分子质量13×103处出现了表达条带(图4),分子质量大小与预期一致,说明GVP蛋白在大肠杆菌BL21(DE3)中成功表达。使用Ni+-NTA树脂纯化目的蛋白,纯化效果较好(使用Quantity One软件分析计算纯度达90%以上)。
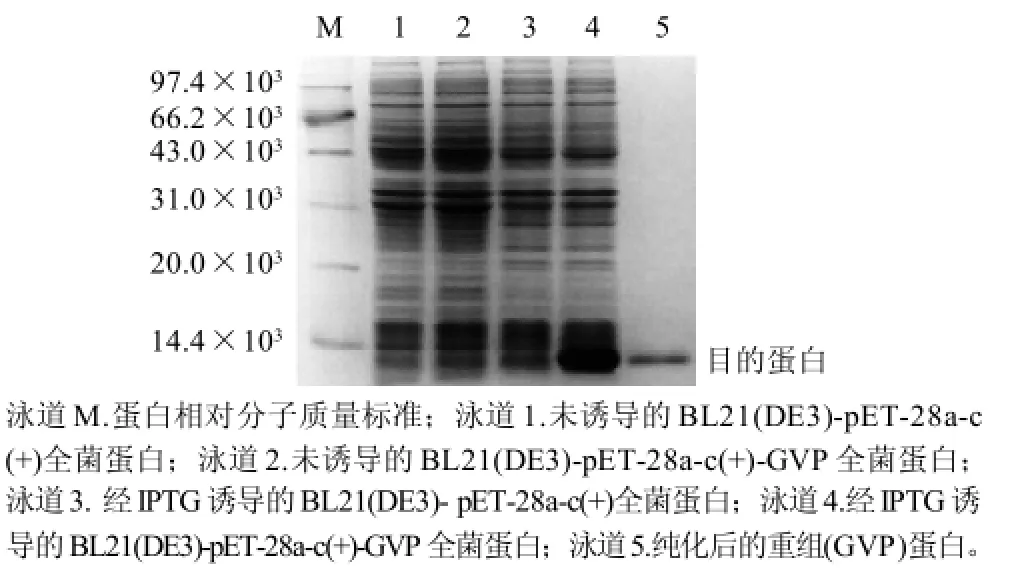
图4 重组蛋白的表达及纯化Fig.4 Expression and purification of recombinant protein
2.3 重组蛋白表达条件的优化
2.3.1 IPTG浓度对GVP蛋白表达的影响
在IPTG浓度分别为0.2、0.4、0.6、0.8、1.0、1.2mmol/L,各诱导4h,发现在0.8mmol/L时,目的蛋白表达量最高(图5)。

图2 第2、3轮重叠延伸PCRFig.2 The second and third steps of overlap extension PCR

图5 诱导剂IPTG浓度对重组蛋白GVP表达的影响Fig.5 Effect of IPTG on the expression of recombinant GVP
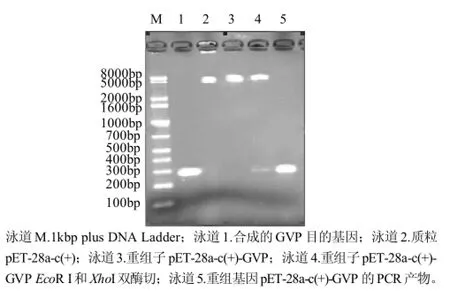
图3 重组基因电泳分析Fig.3 Electrophoretic analysis of recombinant genes
2.3.2 诱导时间对GVP蛋白表达的影响
以0.8mol/L的IPTG分别诱导1、2、4、6、8h,结果显示诱导6h时目的蛋白表达量最大(图6)。

图6 诱导时间对重组蛋白GVP表达的影响Fig.6 Effect of induction time on the expression of recombinant GVP
2.4重组GVP蛋白与单链DNA结合实验结果分析
GVP蛋白与单链DNA结合呈现饱和性,饱和结合量为0.04mol/mol GVP,与双链DNA结合量为0.013mol/mol GVP,结合程度明显低于单链DNA(图7)。根据Scatchard作图法[11],以结合的ssDNA浓度/游离的ssDNA浓度对结合的ssDNA浓度作图,得到一条高度相关直线(图8),由此可计算GVP蛋白与单链DNA结合的解离常数Kd值为7.27×10-5mol/L。
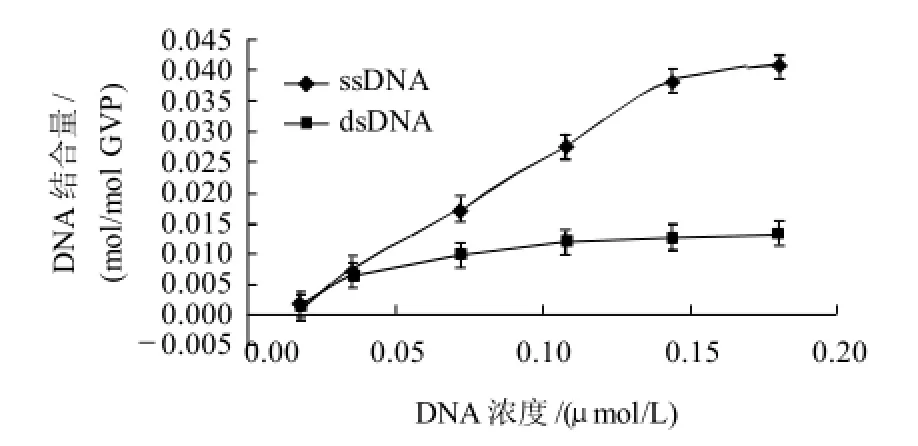
图7 重组GVP与DNA的结合曲线Fig.7 Curve of DNA binding capacity of GVP
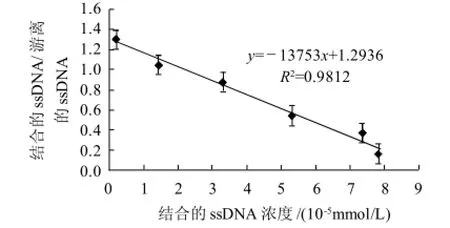
图8 重组GVP与ssDNA结合力的线性关系图Fig.8 Linear relationship between binding ssDNA concentration/free ssDNA concentration ratio and free ssDNA concentration
3 讨 论
在大肠杆菌表达体系中,由于外源蛋白在大肠杆菌中的表达受多种因素的影响,如启动子和终止子的强弱、SD序列、密码子的偏爱性、基因二级结构、宿主菌的遗传背景、培养条件等,其中尤以密码子的偏爱性最为重要[12]。因此为了实现GVP基因在大肠杆菌中的高效表达,采用基因全合成的方式,使用大肠杆菌偏爱密码子替换原密码子,从基因水平上进行优化。
本实验中将GVP基因分成了8段,其中的第1段和第8段为26bp,相对较短,并设计了酶切位点,以便用于扩增全长DNA片段及方便装入载体中。重叠延伸PCR过程中,重叠碱基的数目既决定了每个片段的长度,又决定了扩增的效果,重叠数目过低不利于延伸的正确进行。因此本实验采用每段重叠18bp,以提高重叠延伸PCR的特异性[13-14]。
丝状噬菌体编码的GVP蛋白为87个氨基酸残基,相对分子质量为9682。本实验得到的重组GVP相对分子质量约为13000,主要由目的基因与载体pET-28a(+)中表达氨基末端的一段6×His标签的序列进行融合表达所致,这样即可实现目的蛋白在宿主菌中的高效表达,同时His标签又为后续的重组蛋白分离纯化提供了方便,利用重组蛋白上His标签与Ni+亲和层析柱间高的亲和力,洗涤杂蛋白纯化目的蛋白。
通过DNA结合实验显示出重组GVP蛋白具有较好的单链DNA结合能力,对同一来源的双链DNA亲和作用明显较低。结果表明所得的重组GVP蛋白与天然的GVP蛋白功能相同或相似,属于DNA结合蛋白。
[1]MOU T C, GRAY C W, GRAY D M, et al. The binding affinity of Ff gene 5 protein depends on the nearest-neighbor composition of the ssDNA substrate[J]. Biophysical Journal, 1999, 76(3): 1537-1551.
[2]MOU T C, GRAY C W, TERWILLIGER T C, et al. Ff gene 5 protein has a high binding affinity for single-stranded phosphorothioate DNA[J]. Biochemistry, 2001, 40(7): 2267-2275.
[3]WEN J D, GRAY D M. Selection of genomic sequences that bind tightly to Ff gene 5 protein: primer-free genomic SELEX[J]. Nucleic Acids Research, 2004, 32(22): e182. doi: 10.1093/nar/gnh179.
[4]ZOU Wenquan, ZHENG Jian, GRAY D M, et al. Antibody to DNA detects scrapie but not normal prion protein[J]. PNAS, 2004, 101(5): 1380-1385.
[5]王霞, 李轶女, 赵巧玲, 等. ApNPV编码的DNA 单链结合蛋白基因lef-3的克隆与分析[J]. 蚕业科学, 2005, 31(2): 145-150.
[6]AYRES M D, HOWARD S C, KUZIO J, et al. The complete DNA squence of autographa californica nuclear polyhedrosis virus[J]. Virology, 1994, 202(2): 586-605.
[7]HOROTON R M, PULLEN J K, PEASE L R, et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction [J]. Gene, 1989, 77(1): 51-59.
[8]ZEIDLER M, STEWART G E, BARRACLOUGH C R. et al. New variant Creutzfeldt- Jakob disease: neurological features and diagnostic tests[J]. Lancet, 1997, 350: 903-907.
[9]WILESMITH J W, RYAN J B, HOFFMAN R, et al. Bovine spongiform encephalophathy: epidemiological features 1985 to 1990[J]. Veterinary Record, 1992, 130(5): 90-94.
[10]MOORE J, BOSWELL S, HOFFMAN R, et al. Mutant H-ras overexpression inhibits a random apoptotic nuclease in myeloid leukemia cells[J]. Leukemia Research, 1993, 17 (8): 703-709.
[11]祝庆麟. Scatchard作图及其参数求解法[J]. 中国医学科学院学报, 1986, 8(6): 466-472.
[12]王云龙, 李玲玲, 李晨阳, 等. 猪γ干扰素基因的合成、表达及纯化[J]. 中国生物制品学杂志, 2007, 20(7): 511-514.
[13]陈国梁, 张金文, 陈宗礼, 等. 重叠PCR一步法对三种淀粉合成酶融合基因的构建[J]. 甘肃农业大学学报, 2010, 45(3): 38-43.
[14]符勇耀, 李正国, 李泮志, 等. 优化重叠PCR法进行单链抗体基因扩增和点突变[J]. 生物技术通报, 2009(7): 150-155.
Synthesis, Recombinant Expression and Functional Analysis of Filamentousphage Gene V Protein
ZHANG Da-chuan,FANG Guo-liang,LI Qi,CHEN Jiang-yuan,WANG Lan,LI Rui,LIU Lie-ju,LIU Zhi-guo*
(School of Biology and Pharmaceutical Engineering, Wuhan Polytechnic University, Wuhan 430023, China)
Objective: To explore the gene synthesis, recombinant expression and functions of filamentousphage gene V protein (GVP). Methods: According to the gene sequence of GVP, eight oligonucleotide fragments with the selected E. coli-preferred codon were designed, and the GVP gene sequence was synthesized by overlap extension PCR. Then the synthesized sequence was inserted into pET-28a-c(+) plasmid. The recombinant plasmids obtained were transformed into E. coli to screen positive isolates of GVP. GVP expression in the positive strains was induced with IPTG. The recombinant proteins were purified by Ni+-NTA affinity chromatography. Results: GVP gene was successfully synthesized and highly expressed in E. coli BL21 (DE3) under the induction of IPTG, and the equilibrium dissociation constant was 7.27 × 10-5mol/L between GVP and ssDNA. Conclusion: The recombinant GVP has high affinity with ssDNA and therefore can be used for the ssDNA detection of some pathogenic microorganisms in food.
gene V protein;E. coli BL21 (DE3);recombination;expression
Q782
A
1002-6630(2012)01-0191-04
2011-07-01
武汉市科技局对外科技合作与交流计划项目(201070934341);武汉市科技局现代农业技术创新平台项目(201120637175-5)
张大川(1985—),男,硕士研究生,研究方向为食品安全。E-mail:zhangdachuan1985@163.com
*通信作者:刘志国(1963—),男,教授,博士,研究方向为生物技术与食品安全。E-mail:zhiguo_l@126.com
