牙源性角化囊性瘤的研究进展
2012-03-20刘学恒
贾 黎 刘学恒
北京大学深圳医院口腔科。广东 深圳 518036
牙源性角化囊性瘤(keratocystic odontogenic tumour,KCOT),以往称为牙源性角化囊肿(odontogenic keratocyst, OKC),是一种常见的颌骨牙源性病损,具有较高的增殖潜能,生长方式特殊,保守性手术后易复发[1-3]。2005年WHO对头颈部肿瘤的新分类中,将其归为牙源性良性肿瘤[4]。大部分KCOT为单发病损,部分病例可伴发痣样基底细胞癌综合症(nevoid basal cell carcinoma syndrome,NBCCS)。NBCCS是一种累及多种组织器官的常染体显性遗传病[5],患者可表现多种发育异常,并易患多种类型的肿瘤(详见表1),颌骨多发性KCOT为NBCCS最常见的表现之一,约见于65%~90%的患者。
目前认为KCOT的组织来源为颌骨内的牙板上皮剩余,这些牙齿发育过程中残留的上皮结构受到刺激后发生增殖,继而发生囊肿或肿瘤。然而对于KCOT发病的始动因素及分子机制仍不清楚。NBCCS致病相关基因PTCH1的研究为KCOT的分子发病机制提供了新的理论依据。本文就目前KCOT的研究进展做一综述总结。
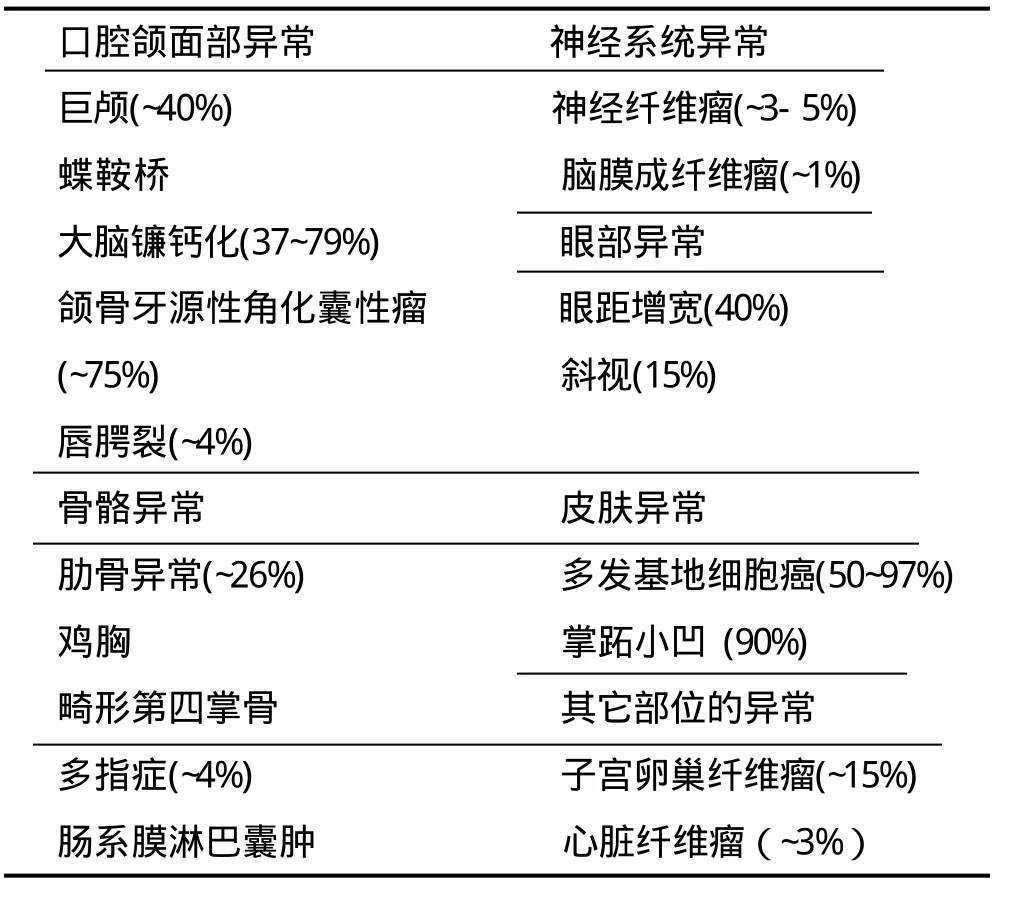
表1 NBCCS的临床表现及外显率[6]
1 KCOT分子发病机制的研究
1.1 KCOT与PTCH1 PTCH1基因全长51kb,位于9q22-31,其编码蛋白为全长1447个氨基酸的12次跨膜蛋白patched。patched作为sonic hedgehog(SHH)的受体,通过Hedgehog信号传导通路对G蛋白偶联受体-smoothened(SMO)进行负调控[7]。在没有SHH配体存在时,patched抑制smoothened,进而抑制下游的信号传导;当SHH与patched结合时,patched对smoothened的抑制作用被解除,Hedgehog通路被激活,其下游的转录因子Gli进入核内,启动下游效应基因的转录,其目的基因包括PTCH1自身,BMP家族,WNT家族、TGFβ家族等。PTCH1的异常失活可导致Hedgehog传导通路的组成性激活,进而引起一系列发育异常和肿瘤发生[8]。
目前认为PTCH1基因是一种肿瘤抑制基因,NBCCS及其相关肿瘤的散发形式可能发生PTCH1基因的突变。我们课题组的系列研究[9-11]和国内外其他研究[12,13]均证实:PTCH1突变不仅频发于NBCCS相关性KCOT,散发的KCOT也可发生PTCH1突变。目前的研究表明,并非所有的KCOT均发生了PTCH1基因的突变,这提示了在KCOT发病机制中,PTCH1基因的失活可能还存在除突变之外还存在其他途径或机制。
1.1.1 PTCH1基因突变 研究表明,NBCCS相关肿瘤及其散发形式视网膜母细胞瘤、基底细胞癌、成神经管细胞瘤等均可发生PTCH1基因突变[14,15,16,17]。对16例综合征相关KCOT和34例散发KCOT进行PTCH1基因的突变检测,分别在14例(87.5%)NBCCS相关KCOT和10例(29.4%)散发性KCOT中检测到26处突变位点(2处突变,c.2619C>A和c.1247C>G发生了两次),其中10处发生移码突变,2处为无义突变,7处错义突变,4处框内插入/缺失,还有3例发生于外显子和内含子接头的突变导致mRNA异常剪切。大部分PTCH1突变(13/26)可导致patched蛋白的提前截断。9/26发生于与hedgehog配体结合相关的两个大的细胞外环上。另一个相对高发的突变位点(5/26)在固醇感受域(sterol-sensing domain,SSD),该功能域在进化上高度保守[18],但功能不明。这些结果结合国内外的研究成果提示:PTCH1基因突变所导致的异常可能与KCOT的发病紧密相关。
1.1.2 PTCH1基因与二次打击理论 肿瘤形成过程包括多个不连续的进程,这些进程可能与特定基因所累积的突变相关。1971年Knudson通过对48例儿童视网膜母细胞瘤发病率进行了回顾性的统计学分析后才提出了“二次打击”理论[19]。该理论认为视网膜母细胞瘤的发生是两次突变的结果。对于双侧患病的家族性患者来讲,第一次基因突变发生于生殖系水平,第二次突变则发生于出生后的体细胞水平;对于单侧患病的散发性视网膜母细胞瘤患者而言,患者往往携带两处体细胞水平的突变。这一现象说明视网膜母细胞瘤的发生是由两次突变事件造成的——即散发肿瘤中的两次体细胞突变,家族性肿瘤中一次体细胞和一次生殖系水平突变。随后,视网膜母细胞瘤的致病基因RB1被克隆,同时发现在肿瘤组织中,RB1两条等位基因由于突变而全部失活[20]。这一观点的提出引发了新的癌症基因分类,癌症相关基因被分为原癌基因和抑癌基因两大类。
Knudson关于肿瘤抑制基因的“二次打击”理论认为[19,21],肿瘤的发生发展需要抑制基因发生两次“打击”事件,才可使该基因的两条等位基因完全破坏,导致基因失活。随着表观遗传学的发展,目前认为造成肿瘤抑制基因的“二次打击”的事件可以为一条等位基因的突变及另一条等位基因的突变、LOH或过甲基化。NBCCS及其相关散发肿瘤的发生被认为是遵循PTCH1的“二次打击”理论的[22,23],即综合征相关的BCCs和KCOTs可能起源于含有遗传性的“第一次打击”的前体细胞,仅需要一次体细胞突变即可引起PTCH1基因的纯合失活,进而引起肿瘤的发生。而KCOT散发形式的发生则需要体细胞水平的两次打击或突变,其中一次可表现为等位基因缺失或启动子的过甲基化。然而目前关于PTCH1基因失活是否遵循经典的“二次打击”模式研究较少,有学者认为PTCH1基因的一次突变打击即可发生肿瘤,即单倍体剂量不足因素。所以目前关于KCOT的发生到底需要一处、两处还是更多PTCH1基因的失活尚无定论;或者并不能用一种统一的模式来解释KCOT的发生。
近年来,通过突变、基因表达异常、体外功能分析及基因敲除小鼠等证据确定了很多候选的肿瘤抑制基因。Knudson的“二次打击”理论已经成为肿瘤抑制基因的检验标准,该理论不仅在RB1等基因中得以证实,在其他肿瘤抑制基因如TP53、WT1等中也均得到了证实。
1997年,Haber和Harlow提出了定义肿瘤抑制基因的严格标准[24],即:这类基因在肿瘤发展过程中发生了突变并失去功能。这个概念的提出试图建立一个可使多数肿瘤抑制基因得以正确纳入的严格标准。和Knudson相同,这个分类也是基于突变的概念之上的,强调突变胜于强调基因的功能。因此,现在很多学者将发生了两处失活打击视为是判定肿瘤抑制基因唯一可靠的证据。然而,这个定义很可能将很多潜在的肿瘤抑制基因排除在外,包括一些通过甲基化等表观遗传学因素失活的基因,或者仅发生了一条等位基因失活的基因,还有一些可诱导肿瘤形成的修饰基因等(这部分基因表现为多态性变异而非突变)。近来年,越来越多的候选肿瘤抑制基因被证实并不遵循经典的“二次打击”模型。
1.1.3 PTCH1基因的表观遗传学研究 表观遗传学因素导致的基因失活是肿瘤发生过程中的重要始动因素之一,研究表明:肿瘤抑制基因启动子区CpG岛可发生过甲基化并导致基因沉默,使蛋白表达水平降低。这种过甲基化可能与基因的杂合性缺失、突变共同构成“二次打击”,导致抑癌基因的两个等位基因全部失活,促使肿瘤发生发展[25]。目前研究已涉及了包括p16、p15、Rb、APC、COX-2等30余种抑癌基因的异常甲基化,其中,启动子区CpG岛的过甲基化几乎在每种类型的肿瘤中都可发生。
目前越来越多通过过甲基化因素失活的基因被报道,故甲基化因素被逐渐认为是肿瘤抑制基因的一个额外的、有效的打击。然而,很多学者始终认为与肿瘤发生相关的截断突变是肿瘤抑制基因的一个确切证据。这一观点长久以来占据统治地位,以至于当一个候选的肿瘤抑制基因如果仅发生了过甲基化而未发生截断突变,就不能被认为是一个真正的肿瘤抑制基因,除非还有其他证据来支持。
迄今仅有少数文献报道PTCH1基因启动子区域的过甲基化疾病发病过程中的作用[26,27,28]:已证实PTCH1基因启动子区域的异常甲基化导致的基因失活参与了乳腺癌、卵巢囊肿及纤维瘤的发病过程,但是在基底细胞癌及成神经管细胞瘤中并未检测到PTCH1基因启动子的过甲基化。这说明PTCH1基因的过甲基化可能与肿瘤的类型有关,KCOT中是否发生了PTCH1基因的过甲基化目前还未见报道,仍有待于深入研究。由于甲基化导致的基因沉默是一个可逆的基因修饰过程,使用甲基化抑制剂可直接恢复抑癌基因功能,因此,改变肿瘤相关基因的甲基化状态很可能为肿瘤治疗提供一个新的途径。
1.2 KCOT与PTCH2 脊椎动物Hedgehog蛋白存在另一个受体——Patched2。人PTCH2基因定位于1p32.3-p32.1,共有22个外显子,编码1 203个氨基酸的12次跨膜蛋白,与PTCH1基因序列有54 % 的同源性。有报道成神经管细胞瘤和BCC中可发生PTCH2基因的突变,最近在1个中国汉族NBCCS家系中,发现致病性PTCH2基因突变(2157G>A)[29]。我们课题组对15例已完成PTCH1基因突变检测的NBCCS相关KCOT进行PTCH2基因的突变筛查[30],发现2例病例携带生殖细胞水平的错义突变,其中1例在核苷酸323位点发生T>C的改变(e.323 T>C),相应的编码氨基酸由亮氨酸变为脯氨酸,该位点位于PTCH2基因的第一个大的细胞外环上,为结合Shh蛋白密切相关的重要结构域。该患者并未检测到PTCH1基因的异常。另1例携带1319位点c>T的改变(e.1319C> T)导致编码氨基酸由丙氨酸变成为缬氨酸,该突变位于PTCH2蛋白的固醇感受域。该患者同时携带一处PTCH1基因的胚系突变为整码插入(e.1338—1339 ins.GCG),即在PTCH1蛋白的第二跨膜区插入了一个丙氨酸,该区域同样位于进化上高度保守的固醇感受域。此外本研究还发现了9处PTCH2的多态性位点,其中3处为尚未报道的新位点(e.2067 T>C,e.2319 G>A,e.3126+14 C>T)。在检测的l5例病例中有13例携带P日,基因突变(13/15),与之相比,NBCCS相关性KCOT患者中PTCH2突变的发生率较低(2/15),且本研究发现在NBCCS患者中,PTCH2的突变可单独存在,亦可与PTCH1的突变同时发生。但迄今为止文献中相关研究较少,PTCH2基因突变发生的频率、类型以及分布特点及其在NBCCS发病中作用机制还有待进一步研究[31]。
1.3 KCOT与SMO 1996年,人们首次分离出了与果蝇同源的鼠和人类的SMO基因,为编码794个氨基酸的连续序列。SMO定位于7q32.3-q34,含有12个外显子,总长为24kb。SMO蛋白属于7次跨膜受体超家族,含有4个糖基化位点,长度为203-205个氨基酸的、半胱氨酸丰富的胞外N-末端(cysteine-rich N-terminal extracellular domain CRD)与跨膜区具有高度保守性,与Wnt家族受体Fizzled相似,提示此区域在SMO功能上的重要作用[32,33,34]。当受到Hh信号刺激时,核内SMO通过磷酸化被转移到细胞表面,变得更加稳定。即使在没有Hh信号的情况下,SMO的过表达也可激活信号的传递,SMO发生激活性突变可导致SMO蛋白聚集于细胞膜,从而激活通路[35,36,37]。
PTCH1的失活性突变使其蛋白丧失对SMO的抑制作用,导致Hedgehog通路的异常激活,这可能是NBCCS患者发生一系列发育异常和肿瘤的重要原因。但并非所有NBCCS相关疾病及其散发病变都有PTCH1基因突变的现象,特别是各种病变的散发形式突变检出率并不高,促使人们进一步探讨Hedgehog信号传导通路中其他重要分子的作用。Xie等[38]报道了3例散发性BCC中两处SMO基因的错义突变,并发现突变的SMO蛋白能够协同腺病毒E1A转化大鼠胚胎成纤维细胞,过表达SMO突变体的转基因鼠皮肤可发生类似于BCC的病损,推测PTCH1的失活突变导致功能丧失和SMO的激活突变导致其功能激活都会导致SHH信号的持续性、非配体依赖性激活,进而导致肿瘤发生。在对16例综合征相关和34例散发性KCOT病变采用PCR—DHPLC筛查和DNA直接测序方法检测SMO基因的突变[9],结果发现3处以往未见报道的单碱基改变,分别位于SMO基因的2号外显子(e.536C>T)、2号内含子(e.IVS537+18C>T)和3号外显子(e.582A>G),经限制性片段长度多态性分析确定为SMO基因的多态性,但并未检测到SMO基因的致病突变。有研究证实:PTCH1对SMO的抑制作用是通过阻止SMO以高磷酸化状态在细胞膜的大量聚集而实现的,外源性SMO的表达可以导致SHH通路目标基因的高表达,提示SMO对SHH信号通路的影响可能是通过对量的调控来实现的[39],而非以其突变体形式。因此进一步明确SMO是否在KCOT发生过程中起作用,还有待于分析SMO在mRNA和蛋白水平上的表达变化[31]。
2 对KCOT间质的研究
上皮-间质相互作用(Epithelial–mesenchymal interactions,EMI)在保持组织稳定方面起关键作用,主要通过细胞外基质(extracellular matrix, ECM)来维持细胞大小、功能和对病理条件下的反应[40]。成纤维细胞,是间质中的主要细胞成分,在邻近的上皮发生肿瘤时也发生改变[41]。生理情况下成纤维细胞可以分泌Ⅰ型胶原和Ⅲ型胶原。胶原是纤维结缔组织的主要有机成分,Ⅰ型胶原纤维含量最多,主要分布在皮肤、肌腱、骨和筋膜内,它常以紧密堆积的形式互相联结,使组织显示出韧性,对张力和拉力有较强的抵抗力;Ⅲ型胶原在组织中的含量较低,但分布广泛,是网状纤维的胶原亚单位。
Junqueira等最早将PSP法应用于组织的胶原检测[42]。PSP法的原理是基于胶原分子丰富的碱性氨基酸和因胶原蛋白平行聚合排列而具有的双折射性质。天狼猩红是强酸性染料,与胶原结合稳定不易褪色,染料分子呈长形展开,在长轴方向以彼此平行的方式附着于每个胶原分子,使胶原的双折射性明显增强,因而入射的正交偏光由于双折射而发生的光干涉更明显,不同折射率表现出不同的干涉色和折射强度。Ⅰ型胶原由多个原纤维粒合成,成熟、粗大,折射率强,在偏光下呈亮黄、橙或红色粗纤维状;Ⅲ型胶原以原纤维形式存在,纤细、幼稚,折射率弱,因此在偏光下为细长的绿色纤维。采用PSP法可在同一视野中观察Ⅰ型和Ⅲ型胶原的形态、分布,并可进行半定量分析。PSP法已用于皮肤病的诊断,牙龈增生和牙源性肿瘤间质的研究[43-46]。
牙源性囊肿和肿瘤都来自牙源性上皮[47],但是它们却表现不同的生物学行为。这些差异一方面来自病损发生过程中上皮成分的特征,另一方面差异则来自病损的间质不同。在以往的OKC中上皮-间质相互作用的研究中,Vedtofte等[48]将OKC上皮移植到裸鼠体内,发现只有在OKC本身的间质支持下,才能保留其典型的组织学表现。这说明OKC的生物学行为同时需要依赖于上皮和间质,间质在调控上皮细胞形态稳定,增殖、分化、粘附等过程起重要作用。研究发现KCOT的侵袭性生长的生物学行为和其特征性间质有关,如KCOT细胞外基质内tenascin和fibronectin表达要高于根尖囊肿(radicular cyst, RC)[49],伴发综合征的KCOT的细胞外基质内tenascin, fibronectin, 基底膜Ⅳ型胶原表达与散发KCOT有明显区别[50]。
Hirshberg等[51]采用PSP法比较OKC与含牙囊肿(dentigerous cyst, DC)和RC的结缔组织基质成分,发现OKC的胶原比较细小、松散,而DC和RC的胶原比较粗大,紧密,分析OKC的间质内可能含前胶原,中间胶原或其它一些病理性胶原成分,认为KCOT的囊壁胶原纤维和牙源性肿瘤的间质相似,提示KCOT的间质不仅是囊壁的支撑者,还在病损的生物学行为中发挥作用。da Silva等[52]用免疫组织化学方法观察到OOC与OKC的囊壁中都含有Ⅰ型胶原和Ⅲ型胶原,OKC囊壁中的胶原纤维排列松散、弥散,没有OOC规律,目前尚无对OOC囊壁中的胶原成分定量分析的研究。本文中采用PSP法分析并首次定量比较OOC和KCOT囊壁中的胶原成分,发现OOC囊壁中主要为Ⅰ型胶原,粗大致密,按一定方向规律排列,形态和分布类似于皮肤和口腔角化粘膜如牙龈[53,54];而KCOT囊壁中主要为Ⅲ型胶原,细小疏松,联结成网状,二者有明显区别。
间质对于保持上皮组织稳定起关键作用,构成细胞复杂的分子交叉传递的外环境。研究表明,间质成分的改变如肌成纤维细胞(myofibroblast,MF)的出现,会导致相邻的上皮发生肿瘤性变化[55,56]。MF具有收缩能力,生理和生化特性与平滑肌细胞相似,由成纤维细胞转化而来,主要促使细胞外基质大量合成并具有促进肿瘤性上皮病损发展的潜能[57,58]。Vered等[59]研究发现OOC囊壁间质中的MF数目显著少于KCOT,KCOT间质中MF数目居然和口腔鳞状细胞癌间质数目没有显著性差异,提示其侵袭性生长行为和MF在间质中表达增高、上皮和间质之间相互作用及产生的相关反应有关。
综上所述,由WHO牙源性肿瘤新分类所引发的有关KCOT是“肿瘤”还是“囊肿”的争论,进一步增加了人们对它的关注程度。对KCOT的保守性手术治疗选择与预防临床复发的问题、KCOT病变微环境中与骨吸收相关的各类分子在颌骨破坏中的作用与调控、致病相关基因细胞遗传学和表观遗传学研究等问题的深入研究将会使我们对KCOT病变本质和发病机制的认识有更加深入的了解。
1.Browne, R.M.. The odontogenic keratocyst. Histological features and their correlation with clinical behaviour [J]. Br Dent J,1971,131(6):249-59.
2.Browne, R.M..The pathogenesis of the odontogenic keratocyst[J]. Proc Int Acad Oral Pathol,1969:28-38.
3.李铁军.牙源性角化囊肿的生长与行为[J]. 中华口腔医学杂志,2000,35(4):306-308.
4.Barnes, L.E., et al. Keratocystic odontogenic tumour. World Health Organization classification of tumours: pathology and genetics of tumours of the head and neck, 2005. Lyon(IARC):330-386.
5.Gorlin, R.J. Nevoid basal cell carcinoma (Gorlin) syndrome [J]. Genet Med,2004,6(6):530-9.
6.High A, Zedan W. Basal cell nevus syndrome[J]. Oncology, 2005,17:160-166.
7.Booth, D.R. The hedgehog signalling pathway and its role in basal cell carcinoma[J]. Cancer Metastasis Rev, 1999, 18(2):261-84.
8.Oldak, M., et al. Clinical aspects of disrupted Hedgehog signaling (Review)[J]. Int J Mol Med,2001,8(4):445-52.
9.Sun, L.S., X.F. Li, and T.J. Li. PTCH1 and SMO gene alterations in keratocystic odontogenic tumours[J]. J Dent Res,2008,87(6):575-9.
10.Li, T.J., et al. PTCH germline mutations in Chinese nevoid basal cell carcinoma syndrome patients[J]. Oral Dis,2008,14(2):174-9.
11.Gu, X.M., et al. PTCH mutations in sporadic and Gorlinsyndrome-related odontogenic keratocysts[J]. J Dent Res, 2006,85(9):859-63.
12.Barreto, D.C., et al. PTCH gene mutations in odontogenic keratocysts[J]. J Dent Res,2000,79(6):1418-22.
13.Ohki, K., et al. PTC gene mutations and expression of SHH, PTC, SMO, and GLI-1 in odontogenic keratocysts[J]. Int J Oral Maxillofac Surg,2004,33(6):584-92.
14.Raffel, C., et al. Sporadic medulloblastomas contain PTCH mutations[J]. Cancer Res,1997,57(5):842-5.
15.Soufir, N., et al. PTCH mutations and deletions in patients with typical nevoid basal cell carcinoma syndrome and in patients with a suspected genetic predisposition to basal cell carcinoma: a French study[J]. Br J Cancer, 2006,95(4):548-53.
16.Savino, M., et al. Spectrum of PTCH mutations in Italian nevoid basal cell-carcinoma syndrome patients: identification of thirteen novel alleles[J]. Hum Mutat, 2004,24(5):441.
17.Ping, X.L., et al. PTCH mutations in squamous cell carcinoma of the skin[J]. J Invest Dermatol,2001,116 (4):614-6.
18.Ohgami, N., et al. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain[J]. Proc Natl Acad Sci U S A,2004,101(34):12473-8.
19.Knudson, A.G., Jr., Mutation and cancer: statistical study of retinoblastoma[J]. Proceedings of the National Academy of Sciences of the United States of America, 1971,68(4):820-3.
20.Friend, S.H., et al. A Human DNA Segment with Properties of the Gene That Predisposes to Retinoblastoma and Osteosarcoma[J]. Nature,1986,323(6089):643-646.
21.Knudson, A.G. Hereditary cancer: two hits revisited[J]. Journal of Cancer Research & Clinical Oncology,1996, 122(3):135-40.
22.Levanat, S., et al. A two-hit model for developmental defects in Gorlin syndrome[J]. Nat Genet,1996,12(1):85-7.
23.Koch, C.A., et al. Two-hit model for tumourigenesis of nevoid basal cell carcinoma (Gorlin) syndrome-associated hepatic mesenchymal tumour[J]. Am J Med Genet,2002,109 (1):74-6.
24.Haber, D. and E. Harlow. Tumour-suppressor genes: evolving definitions in the genomic age[J]. Nature Genetics, 1997,16(4):320-322.
25.Jones, P.A. and P.W. Laird. Cancer epigenetics comes of age[J]. Nature Genetics,1999,21(2):163-7.
26.Wolf, I., et al. Unmasking of epigenetically silenced genes reveals DNA promoter methylation and reduced expression of PTCH in breast cancer[J]. Breast Cancer Research & Treatment,2007,105(2):139-55.
27.Cretnik, M., et al. The Patched gene is epigenetically regulated in ovarian dermoids and fibromas, but not in basocellular carcinomas[J]. International Journal of Molecular Medicine,2007,19(6):875-83.
28.Pritchard, J.I. and J.M. Olson. Methylation of PTCH1, the Patched-1 gene, in a panel of primary medulloblastomas [J]. Cancer Genetics & Cytogenetics,2008,180(1):47-50.
29.Fan Zhipeng,Li Jingyuan,Du Juan,et a1.A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family[J].J Med Genet,2008,45(5):303-308.
30.徐丽莉,李铁军.伴痣样基底细胞癌综合征的牙源性角化囊性瘤中PTCH2基因的突变检测[J].北京大学学报:医学版,2008,40(1):15-18.
31.李铁军,孙丽莎,罗海燕,等. 颌骨牙源性角化囊性瘤的研究[J]. 北京大学学报:医学版,2009,41(1):16-20.
32.Stone DM. et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog[J]. Nature 1996,384:129-134.
33.Marigo V, Davey RA, Zuo Y, Cunningham, J. M. & Tabin, C. J. Biochemical evidence that patched is the Hedgehog receptor[J]. Nature 1996,384:176-179.
34.Nusse R. Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface[J]. Development,2003,130:5297-5305.
35.Denef N, Neubuser D, Perez L. & Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened[J]. Cell,2000, 102:521-531.
36.Zhang C, Williams EH., Guo Y, Lum L & Beachy PA. Extensive phosphorylation of Smoothened in Hedgehog pathway activation[J]. Proc. Natl Acad. Sci USA,2004, 101:17900-17907.
37.Denef N, Neubuser D, Perez L & Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened[J]. Cell,2000,102:521-531.
38.Xie Jingwu,Murone M,Luoh SM,et a1.Activating Smoothened mutations in sporadic basal—cell carcinoma [J].Nature,1998,391(6662):90-92.
39.Hynes M,Ye Weilan,Wang Kevin,et a1.The seven—transmembrane receptor smoothened cell--autonomously induces multiple ventral cell types[J].Nat Neurosci,2000,3(1):41-46.
40.Tlsty, T.D. and P.W. Hein. Know thy neighbor: stromal cells can contribute oncogenic signals[J]. Curr Opin Genet Dev,2001,11(1):54-9.
41.Bergers, G. and L.M. Coussens. Extrinsic regulators of epithelial tumour progression: metalloproteinases[J]. Curr Opin Genet Dev,2000.,10(1):120-7.
42.Junqueira, L.C., W. Cossermelli, and R. Brentani. Differential staining of collagens type I, II and III by Sirius Red and polarization microscopy[J]. Arch Histol Jpn,1978,41(3):267-74.
43.Trau, H., D. Dayan, A. Hirschberg, et al. Connective tissue nevi collagens. Study with picrosirius red and polarizing microscopy[J]. Am J Dermatopathol,1991,13 (4):374-7.
44.Dayan, D., T. Waner, H. Tal, et al. Polarization microscopy of picrosirius red-stained collagen from oxodipineinduced hyperplastic gingiva of beagle dogs[J]. Int J Exp Pathol,1993,74(3):225-8.
45.Nyska, A. and D. Dayan. Ameloblastic fibroma in a young cat[J]. J Oral Pathol Med,1995,24(5):233-6.
46.Hirschberg, A., A. Buchner, and D. Dayan. The central odontogenic fibroma and the hyperplastic dental follicle: study with Picrosirius red and polarizing microscopy [J]. J Oral Pathol Med,1996,25(3):125-7.
47.Sapp, J., P., L. Eversole, R., G. Wysocki, P., et al. Contemporary oral and maxillofacial pathology[J]. 2nd ed. St. Louis: Elsevier,2004:35,135-6.
48.Vedtofte, P., P. Holmstrup, and E. Dabelsteen. Human odontogenic keratocyst transplants in nude mice[J]. Scand J Dent Res,1982,90(4):306-14.
49.de Oliveira, M.D., J.L. de Miranda, R.F. de Amorim, et al. Tenascin and fibronectin expression in odontogenic cysts[J]. J Oral Pathol Med,2004,33(6):354-9.
50.Amorim, R.F., G.P. Godoy, H.C. Galvao, et al. Immunohistochemical assessment of extracellular matrix components in syndrome and non-syndrome odontogenic keratocysts[J]. Oral Dis,2004,10(5):265-70.
51.Hirshberg, A., S. Sherman, A. Buchner, et al. Collagen fibres in the wall of odontogenic keratocysts: a study with picrosirius red and polarizing microscopy[J]. J Oral Pathol Med,1999,28(9):410-2.
52.da Silva, M.J., S.O. de Sousa, L. Correa, et al. Immunohistochemical study of the orthokeratinized odontogenic cyst: a comparison with the odontogenic keratocyst[J]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod,2002,94(6):732-7.
53.Kurkinen, M., A. Vaheri, P.J. Roberts, et al. Sequential appearance of fibronectin and collagen in experimental granulation tissue[J]. Lab Invest,1980,43(1):47-51.
54.Abrahao, I.J., M.D. Martins, E. Katayama, et al. Collagen analysis in human tooth germ papillae[J]. Braz Dent J, 2006,17(3):208-12.
55.Mareel, M. and A. Leroy. Clinical, cellular, and molecular aspects of cancer invasion[J]. Physiol Rev,2003,83(2): 337-76.
56.De Wever, O. and M. Mareel. Role of myofibroblasts at the invasion front[J]. Biol Chem,2002,383(1):55-67.
57.Tomasek, J.J., G. Gabbiani, B. Hinz, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling [J]. Nat Rev Mol Cell Biol,2002,3(5):349-63.
58.De Wever, O. and M. Mareel. Role of tissue stroma in cancer cell invasion[J]. J Pathol,2003,200(4):429-47.
59.Vered, M., I. Shohat, A. Buchner, et al. Myofibroblasts in stroma of odontogenic cysts and tumours can contribute to variations in the biological behavior of lesions[J]. Oral Oncol,2005,41(10):1028-33.
