鼠尾胶原蛋白提取、分离、纯化方法的建立及鉴定
2012-02-03任海涛钟志勇郑佳琳饶子亮邝少松唐小江
任海涛,钟志勇,郑佳琳,饶子亮,邝少松,王 刚,唐小江
(广东省医学实验动物中心,佛山 528248)
鼠尾胶原蛋白是胶原蛋白的一种,主要为Ⅰ型胶原蛋白[1]。Ⅰ型胶原蛋白主要存在于真皮、腱、骨、牙本质,以纤维形式形成胶原,由三股缠绕形成多肽链组[2]。作为一种新型生物材料,鼠尾胶原蛋白并不为众人所知,但是其具有的诸多优势,如低抗原性、生物可降解性、优越的生物相容性并有利于细胞贴附和迁移等特点[3],为细胞增殖和功能分化提供了合适的微环境[4-5],且它来源广泛,成分单一,制备比较方便,是开发具有相关功能性生物医疗器械的好材料[6]。
目前,国内外对胶原蛋白提取、分离、纯化主要有中性盐法[7]、酸提取法[8-9]、碱提取法[6-10]、酶法[11-12]等,但中性盐法、酸提取法、碱提取法方法均存在着提取得率低,且操作复杂,还可能破坏胶原蛋白的三维立体结构[13]。酶法应用较广,但仍存在着纯化困难的问题。迄今为止,还没有针对鼠尾胶原蛋白进行大规模制备的相关研究的报道。
本研究的目的就是为了克服现有制备鼠尾胶原蛋白方法所存在的提取得率低、分离纯化工艺复杂、没有大规模生产方法等缺点,提供一种工艺简单、提取得率和纯度高的鼠尾胶原蛋白的制备方法。为大量获得鼠尾胶原蛋白并进行更深层次的功效研究提供理论支持和实践基础。
1 材料和方法
1.1 实验动物
SD大鼠鼠尾,由广东省医学实验动物中心提供[生产许可证号SCXK(粤)2008-0002;使用许可证号SYXK(粤)2008-0002]。
1.2 仪器与试剂
835-50型氨裁酸自动分析仪(日本Hitachi公司);J26XP大型冷冻离心机(美国Beckman Coulter公司);Mini-PROTEAN Tetra Cell电泳仪(美国BIORAD公司);130型制冰机(宁波新芝公司); MIKRO20离心机(德国Hettich公司);万分之一天平(德国Sartorius公司);RH basic 2磁力搅拌器(德国IKA公司);SRM-CD47医用保存柜(日本Sanyo公司)。
胃蛋白酶(Sigma公司);醋酸、氯化钠(广州中南化工公司)、万级透析袋(广州威佳公司);Tris-HCl盐、实验用水为18.2 mΩ超纯水;其它化学试剂均为国产分析纯。
1.3 实验方法
本研究所采用的新型“酸混合酶”法通过对提取、分离、纯化鼠尾胶原蛋白的单因素参数进行优化,得到了更优的扩大化生产工艺参数。首先,剥取SD大鼠尾腱需按照“从细到粗”的原则,在冰浴上进行,避免鼠尾胶原蛋白变性。同时,应将鼠尾软骨在剥离前先自行拧成半断裂状,这样可以最大限度的剥取鼠尾腱。再次,对剥离的白色鼠尾腱应立即放入含有0.05 mol/L Tris-HCl缓冲盐的1 mol/ L NaCl溶液中处理24 h。以除去可溶性多糖、血液及其他杂质等。提取鼠尾胶原蛋白的步骤如下:1.使用溶解有一定比例胃蛋白酶的酸溶液对初步处理的鼠尾腱进行酶解,持续处理一段时间待混合物变成无色、透明、粘稠液体后即可在4℃,5000 g离心力下离心20 m in,收集上清即为粗制鼠尾胶原蛋白原液。2.将一定浓度的氯化钠溶液加入其中,持续搅拌直至有白色絮状沉淀从溶液中析出,继续加入氯化钠溶液至沉淀不在析出为止。4℃,5000 g离心力下离心20 m in后获得白色沉淀。沉淀物加入一定浓度的酸溶液溶解。3、将溶解后的鼠尾胶原蛋白溶液继续反复进行盐析处理,重复步骤2的过程2~4次。持续在超纯水中透析3 d,每天换液3次以除去鼠尾胶原溶液中的无机盐类,获得的鼠尾胶原蛋白经SDS-PAGE蛋白质电泳、氨基酸含量分析仪的鉴定,纯度好。4、通过建立起来的提取方法,继续对提取单因素进行了优化:
1.3.1 所用酸溶液的确定
不同酸溶液可以对酶解产生不同的影响,比较稀盐酸、醋酸、柠檬酸这3种酸溶液用于提取鼠尾胶原蛋白,每种酸溶液加入用于提取的鼠尾腱1 g,其他实验条件一致。3种酸溶液浓度均为0.05 mol/ L,持续4℃搅拌72 h。最终在0.05 mol/L浓度下的稀盐酸、醋酸、柠檬酸提取条件下,分别获得鼠尾胶原0.27 g、0.43 g、0.38 g。故选用醋酸溶液作为提取参数条件。
1.3.2 所用酸溶液浓度的确定
在2.2.1的优化前提下,继续对提取所用的酸溶液浓度进行了优化,选用了0.025 mol/L、0.05 mol/L、0.10 mol/L这3个浓度。每个浓度酸溶液加入用于提取的鼠尾腱1 g,其他实验条件一致。分别获得鼠尾胶原0.37 g、0.44 g、0.38 g。故选用0.05 mol/L浓度作为提取参数条件。
1.3.3 所用酶溶液浓度的确定
在酸溶液中胃蛋白酶可以发挥良好的酶解效果,但综合成本和得率考虑需要对酶溶液浓度进行探讨。本研究使用1∶10000胃蛋白酶进行试验,使用配比分别为于50 m L 0.05 mol/L醋酸溶液溶解10 mg、50 mg、100 mg、200 mg,加入用于提取的鼠尾腱1 g,其他实验条件一致。分别获得鼠尾胶原0.27 g、0.34 g、0.44 g、0.45 g。考虑到200 mg没用量并没有大幅度提供得率,故选用100 mg胃蛋白酶/50m L 0.05 mol/L醋酸溶液浓度(1∶500)作为提取参数条件。
1.3.4 所用酶作用时间的确定
时间是大规模生产上必须要考虑的一个问题,本研究优化了酶解时间,分别选用酶解36 h、48 h、72 h、96 h,其他实验条件一致。每个提取时间加入用于提取的鼠尾腱1 g,获得鼠尾胶原蛋白0.15 g、0.36 g、0.46 g、0.45 g。故选用酶解72 h作为提取参数条件。
1.3.5 所用氯化钠浓度的确定
优化了分级盐析所用的氯化钠浓度,分别选用1 mol/L、2 mol/L、4 mol/L,其他实验条件一致。每个提取时间配置用于提取的鼠尾腱1 g,获得鼠尾胶原蛋白0.35 g、0.45 g、0.45 g。故选用2 mol/L作为提取参数条件。
2 结果
通过单因素条件优化,获得的最佳提取、分离、纯化条件为:0.05 mol/L的醋酸,1∶500酶用量、酶解72 h、2 mol/L氯化钠溶液分级盐析。提高了提取鼠尾胶原蛋白的得率,为大批量生产鼠尾胶原蛋白打下了实践基础,提供了理论支持。经SDS-PAGE电泳和氨基酸含量分析的鉴定,获得的鼠尾胶原蛋白纯度高。本研究简化了提取工艺,每条大鼠鼠尾(总重)平均约5 g,可以提取到干重为200 mg的鼠尾胶原蛋白,提取得率约为4%左右。较传统的方法提取得率仅为0.2%,提取得率提高了20倍以上。

图1 鼠尾胶原蛋白电泳图Fig.1 Protein electrophoresis of rat tail collagen
电泳图中,其中1~4道为5 mg/m L浓度,1、2道为SIGMA公司产品,3、4道为自制鼠尾胶原蛋白。5、6道为20 mg/m L浓度自制鼠尾胶原蛋白,7、8道为2.5 mg/m L浓度自制鼠尾胶原蛋白。
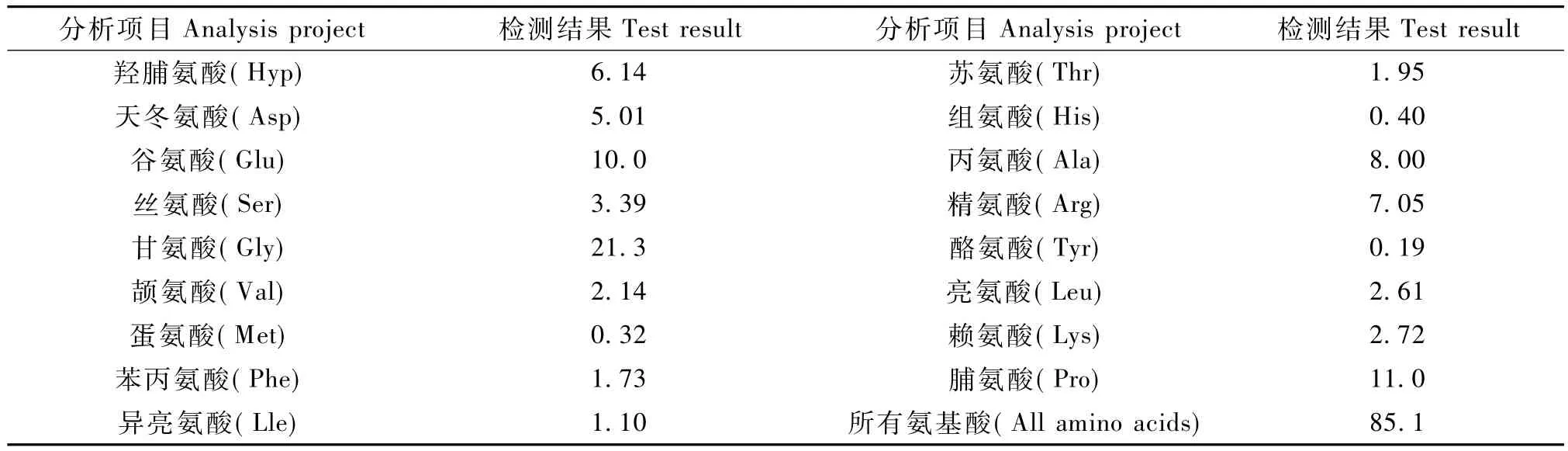
表1 鼠尾胶原蛋白氨基酸含量分析表Tab.1 Content analysis chart of amino acid in rat tail collagen
3 讨论
传统的提取鼠尾胶原蛋白的方法主要是中性盐法、酸提取法、碱提取法、酶法等,但是均存在着不同程度的缺点。酸提取法提取迅速,但色氨酸全部被破坏,丝氨酸和络氨酸部分被破坏;碱提取法造成胶原肽键被水解,含羟基和巯基的氨基酸全部被破坏且产生消旋;中性盐法造成肽链间的离子键会被盐打开造成吸水膨胀,同时扩大生产的工艺不稳定;同时以上3种方法还存在着得率低的共同缺点,制约了鼠尾胶原蛋白的大规模提取。酶法反应条件温和,能保持胶原的三股螺旋结构,但水解又不够彻底。本研究所采用的新型“酸混合酶”法结合了酸提取法和酶法的优势,解决了单一方法提取过程中存在的固有缺陷,同时通过对提取、分离、纯化鼠尾胶原蛋白的单因素参数进行优化,使提取得率较传统工艺提高了20倍以上。确定了工艺简单、操作方便、提取得率高的鼠尾胶原蛋白提取、分离、纯化方法。
本研究所提取的鼠尾胶原蛋白经过鉴定:(1) SDS-聚丙烯酰胺凝胶电泳鉴定,其制备的鼠尾胶原蛋白与现有SIMGA公司的商业化鼠尾胶原蛋白进行了对比,两者图谱无差异。从电泳结果上可以看出,制备的鼠尾胶原蛋白质量高,纯度好。其电泳结果一致。(2)样品经水解,经高压液相色谱检验(样品处理按GB/T 5009.124-2003水解,检测方法按JY/T 024-1996),采用氨基酸分析仪,经广州市分析检测中心对本研究中制备的鼠尾胶原蛋白进行氨基酸含量分析表明,其含有①胶原蛋白特征氨基酸羟脯氨酸,含量约为8%左右;②不含胱氨酸、色氨酸这两种氨基酸,与其自身特征相符;③甘氨酸含量接近总氨基酸含量的三分之一,与其自身特征相符。说明制备的鼠尾胶原蛋白质量高,达电泳纯。为接下来鼠尾胶原蛋白的扩大化生产提供了合适的工艺参数,为大量获得鼠尾胶原蛋白这种生物可吸收材料并进行更深层次的功效方面研究提供了理论支持和实践基础。
[1]张铁良,屠军波,杨壮群等.以鼠尾胶原蛋白为支架的真皮类似物的建立[J].中国美容医学,2006,2(15):131-134.
[2]李艳佳,焦建丽,张佃财等.在铺有鼠尾胶原的TransWell培养板套皿气-液交界面培养角质形成细胞形成皮肤样器官[J].中国组织工程研究与临床康复.2009,13(42):8211 -8215.
[3]胡莹莹,蒋宁一,刘雄英等.鼠尾胶原三维诱导E14小鼠胚胎干细胞分化为甲状腺类似细胞的初步研究[J].广东医学.2009,3(30):327-330.
[4]Vailhe B,Vittet D,Feige JJ.In vitro models of vasculogenesis and angiogenesis[J].Lab Invest,2001,81(4):439-452.
[5]Montanez E,Casaroli-Marano RP,Vilaro S.et al.Comparative study of tube assembly in three-dimensional collagen matrix and on Matrigel coats[J].Angiogenesis,2002,5(3):167-172.
[6]Bell E,Ivarsson B,Merribl C.Production of a tissue-like structure by contraction of collagen lattices by human fibroblasts of different proliferative potential in vitro[J].Proc Natl Acad Sci,1979,76(3):1274-1278.
[7]Fielding A M.Preparation of neutral salt soluble collagen[J].The Methodology of connective Tissue Research Joynson-Bruvvers,Oxford,1976:9-12.
[8]Sajithlal GB,Chithra P,Chandrakasan G.Effect of curcumin on the advanced glycation and cross-linking of collagen in diabet in rats[J].Biochem l Pharmacol,1998,56(12):1607.
[9]杨志明.组织工程(第1版)[M].北京.化学工业出版社,2002:217.
[10]Schor SL,Schor AM,Winn B.et al.The use of three dimensional collagen gels for the study of tumour cell invasion in vitro:experimental parameters influencing cellm igration into the gelmatrix[J].Int JCancer,1982,15;29(1):57-62.
[11]Kochakian M,Majula BN,Egan JJ.Chronic dosing with amino guanidine and novel advanced glycosylation and product formation inhibitors ameliorates cross-linking of tail tendon collagen in STZ-induced diabetic rats[J].Diabet es,1996,45(12):1694.
[12]Oturai PS,Christ ensen M,Rolin B,et al.Effects of advanced glycation End-product inhibition and cross-link breackage in diabetic rats[J].Metabolism,2000,49(8):996.
[13]张文熊,李欣,魏娜等.酶法提取胶原的研究[J].中国皮革,2006,12(23):15-17.
