巨噬细胞与肾间质纤维化
2012-02-01安惠霞张庆芳
陈 晓 崔 艳 安惠霞 张庆芳
(哈尔滨医科大学附属第二医院肾内科二病区,黑龙江 哈尔滨 150086)
巨噬细胞与肾间质纤维化
陈 晓 崔 艳 安惠霞 张庆芳
(哈尔滨医科大学附属第二医院肾内科二病区,黑龙江 哈尔滨 150086)
肾间质纤维化;巨噬细胞
肾间质纤维化是慢性进展性肾病的重要组织学表现,是导致终末期肾衰竭的最终共同通道,其病变程度与慢性肾脏病预后密切相关〔1〕。动物和临床试验表明,各种肾脏疾病进行性肾功能恶化主要取决于肾间质损伤严重程度。传统观点认为肾间质纤维化发病机制主要是:各种致病因子如炎症、损伤、药物、糖尿病和遗传因素等导致肾脏损伤后,肾固有细胞活化,释放化学趋化因子,致使循环中炎症细胞浸润肾间质,进一步产生和分泌促炎症和纤维化的细胞因子;介导炎症反应的级联放大与持续,同时使肾组织局部产生大量氧自由基及碎片组织,引起肾小管细胞凋亡,肾小管萎缩。在细胞因子作用下肾小管上皮-间充质细胞转分化(EMT),成纤维细胞活化,胶原分泌增加而降解减少,造成间质纤维化。可见炎症细胞浸润是肾间质纤维化形成过程中的重要环节〔2〕。而在各种肾小球肾炎中,间质浸润的炎症细胞以单核/巨噬细胞为主〔3〕,因此巨噬细胞浸润与肾脏慢性炎症和纤维化有密切关系。本文对两者间关系及研究现状作一综述。
1 巨噬细胞
巨噬细胞是单核细胞随血流到全身各种组织后分化而成,是单核/吞噬细胞系统中高度分化、成熟的细胞类型,所在部位不同类型和命名也不一样。巨噬细胞可分泌释放血小板源性生长因子(PDGF)、一氧化氮(NO)、活性氧(ROS)、白细胞介素(IL)、肿瘤坏死因子(TNF)-α、补体和金属蛋白酶类等〔4〕,参与局部炎症反应,发挥吞噬防御和抗原提呈功能。基于细胞表面标志和细胞因子分布不同,将巨噬细胞分为两种表型(如图1):M1型和 M2型。M1型通过经典途径活化,γ-干扰素(IFN-γ)、脂多糖(LPS)或细胞因子〔如 TNF和粒细胞-巨噬细胞集落刺激因子(GM-CSF)〕能诱导经典的M1巨噬细胞活化,产生和分泌 IL-1,6,12,TNF-α,巨噬细胞趋化蛋白(MCP)-1,CC亚族趋化因子(CCL2,CCL3,CCL4,CCL20/MIP-3a),ROS,NO等细胞因子,是导致炎症和免疫损伤的主要成分。M2型是经选择性活化,IL-4、IL-13、免疫复合物、IL-10等多种刺激物诱导的活化,产生分泌纤维连接蛋白(FN)、galectin-3(B-半乳糖苷黏合素家族成员之一)、TNF-α、IL-10,12、胰岛素样生长因子(IGF)、转化生长因子(TGF)-β、细胞外基质(ECM)等,具有强的吞噬能力,可产生营养因子,减轻促炎症因子分泌〔5〕。
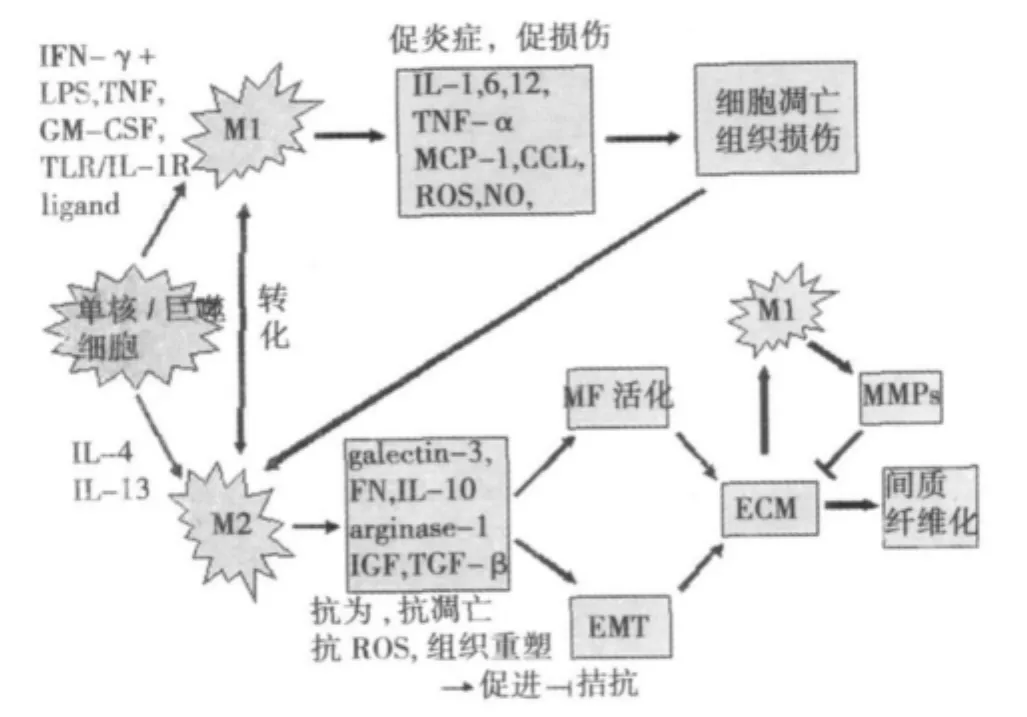
图1 不同表型巨噬细胞与肾脏间质纤维化关系
2 巨噬细胞浸润的机制
目前肾组织中巨噬细胞浸润机制不完全清楚,但目前认为:在各种炎症因子、黏附分子、趋化因子共同作用下,血液中单核细胞与血管内皮细胞发生黏附,同时诱导趋化因子及慢性炎症因子等大量释放,导致肾间质单核细胞局部广泛聚集,分化为巨噬细胞。肾组织局部化学趋化因子和黏附分子表达增加,在巨噬细胞局部组织聚集过程中起重要作用。而炎症因子是介导炎症放大和持续的基础。
2.1 细胞黏附分子 细胞黏附分子是细胞间信息沟通的物质基础,包括选择素,血管细胞黏附分子(VCAM)-1,细胞间黏附分子(ICAM)-1及巨噬细胞抑制因子(MIF)。选择素是细胞黏附分子家族主要成员,其与配体介导循环中炎症细胞与血管内皮细胞的联系,导致炎症细胞捕获与移动。选择素有3种:E-、P-和L-选择素。单侧输尿管梗阻(UUO)模型是研究肾小管间质纤维化的经典模型。在敲除3种选择素(EPL-/-)的鼠UUO模型中,不仅减少巨噬细胞间质浸润,且减少胶原沉积和小管细胞凋亡〔6〕。MIF是一种垂体前叶及外周巨噬细胞产生的蛋白质。MIF能活化巨噬细胞,抑制其游走移动,增强其黏附、吞噬作用,调节白细胞活性和成纤维细胞增殖。VCAM-1,ICAM-1两者在促进固有炎症细胞黏附到血管壁过程中起重要作用,静脉注射反转录核苷酸来对抗ICAM-1基因表达,可减轻UUO术后巨噬细胞浸润及ECM产生〔7〕。
2.2 炎症趋化因子 单核/巨噬细胞炎症趋化因子由巨噬细胞及内皮细胞等多种细胞产生,包括 CXC,CC亚族,C亚族MCP-1等,其主要功能是趋化和激活单核细胞至炎症部位。在UUO模型中两种的表达从第2~10天逐渐增加〔8〕。体内封闭MCP-1基因,可减轻UUO术后巨噬细胞浸润及间质纤维化〔9〕。删除CC受体(CCR)2(CC受体2)基因或应用CCR2阻滞剂也可减轻巨噬细胞浸润及间质纤维化〔10〕。
2.3 炎症因子 炎症细胞激活涉及许多细胞因子,某些因子同时又可被炎症细胞以自分泌方式进行分泌。一些因子如IL-1、2、6、8、10、12、17、18 和 TNF-α 等促进巨噬细胞炎症特征,放大炎症反应。另外如 IL-4、11、13抑制该特性。IL-1和TNF-α作为重要的急性期炎症介质在梗阻肾炎症细胞浸润中起主要作用〔11,12〕。单核/巨噬细胞是 IL-1主要来源,在 UUO 模型中给予IL-1受体拮抗剂抑制活性后,不仅减轻巨噬细胞间质浸润,且减少了ICAM-1和α-肌动蛋白(肌成纤维细胞的标志物)表达〔11〕。TNF-α是一种单核因子,主要由单核细胞和巨噬细胞产生,TNF-α在UUO早期就产生,在UUO术后3 d达到高峰,其后低水平持续存在。TNF-α可刺激单核巨噬细胞、肾小球内皮细胞、成纤维细胞和肾小管上皮细胞大量产生IL-1、8。TNF-α也能部分介导炎症细胞浸润和肾小管细胞凋亡,刺激胶原产生和成纤维细胞增殖〔12〕。骨化醇可通过减少TNF-α表达减轻巨噬细胞及T淋巴细胞浸润〔13〕。
3 巨噬细胞促进间质纤维化
在免疫复合物大鼠肾炎模型(类似于膜增生性肾炎)中,在造模后14 d清除巨噬细胞也可减轻肾小管萎缩和肾间质纤维化〔14〕。而在非免疫复合物模型,如UUO中,在术后10 d清除巨噬细胞也同样减轻肾间质纤维化〔15〕,因此,肾间质纤维化可能与免疫复合物无直接关系,而是继发于如巨噬细胞浸润引起的间质损伤〔16〕。
大量研究报道,浸润的巨噬细胞数量与纤维化程度呈正比,敲除巨噬细胞基因或抑制负责巨噬细胞再生的蛋白编码基因,可使肾纤维化程度减轻〔17,18〕。巨噬细胞本身也能产生大量致纤维化因子 TGF-β1、结缔组织生长因子(CTGF)〔19〕,合成、分泌FN和Ⅰ型胶原,硫酸软骨素等ECM〔20〕,巨噬细胞分泌的galectin-3是引起TGF-β1介导成纤维细胞活化和ECM合成的主要机制〔21〕。巨噬细胞也可分化为成纤维细胞,促进纤维化形成〔22〕。在新生大鼠 UUO模型中,早期通过 CCR-1拮抗剂BX471阻止白细胞募集反应,可减少单核细胞浸润及肾小管上皮细胞间充质转分化,减轻肾小管上皮细胞凋亡和间质纤维化〔23〕。靶向性抑制巨噬细胞肾组织募集和活化,可改善炎症和纤维化〔24〕。
4 部分巨噬细胞抗纤维化作用
巨噬细胞不仅介导组织损伤,也参与组织修复,炎症控制及抗纤维化过程。Nishida等〔25〕发现去除小鼠体内巨噬细胞,在UUO后14 d纤维化程度显著增加,而体外输注巨噬细胞,肾间质局部巨噬细胞数目显著增加,间质纤维化减轻。纵向研究发现:在UUO模型中,间质巨噬细胞增加呈双向性,在术后的第一个24 h间质巨噬细胞急剧增加达到第一个高峰,第二个高峰出现在之后的72 h,在后期(术后14 d)间质巨噬细胞的数量与纤维化程度呈负相关〔26〕。
在对巨噬细胞表型研究发现:不同表型的巨噬细胞在炎症及纤维化中的作用不同(图1)。M1型巨噬细胞分泌的细胞因子如 IL-1β,TNF-α,IL-12,NOS2,ROS,IL-6,CCL17 等对组织有损伤作用。M1型自身并促使肌成纤维细胞产生基质金属蛋白酶(MMPs)〔26〕,而MMPs可降解ECM,起抗纤维化作用。M2型巨噬细胞促血管生成、细胞增殖和ECM重塑〔27〕,可见M2型巨噬细胞具有组织修复作用。在UUO术后10 d解除梗阻,间质巨噬细胞浸润和纤维化减轻,且解除梗阻后第7天以CD204和CD206为代表的 M2型细胞比值增加〔28〕,预示着组织修复开始。组织损伤和细胞凋亡(主要有M1介导)可活化M2型巨噬细胞,吞噬碎片和分泌生长因子,修复损伤〔29〕,而M2型巨噬细胞又可产生大量TGF-β1,可促进肾纤维化〔26〕。M1型早期产生MMPs可降解基底膜成分,促炎症细胞间质迁移,但在后期由于ECM沉积增加,刺激M1型继续产生MMPs,降解ECM,起抗纤维化作用。
巨噬细胞也不是固定在某一特定的活化状态。在某些特定条件下,它们可以从一种状态迅速转到另一种状态。随着凋亡细胞的吞噬,经典活化的M1型可转化为M2型〔30〕。肿瘤相关巨噬细胞(TAMs)是一种与M2相似的巨噬细胞,它可在抗炎与抑制免疫不同功能表型之间相互转化,这种转化取决于肿瘤位置和病情进展〔31〕。这为有目的地促进巨噬细胞表型转化来达到抗炎抗纤维化作用提供了思路。
巨噬细胞也可分泌有修复保护作用的成分〔血管形成素(Ang)1,肝细胞生长因子(HGF)〕。例如:在肾脏,HGF是公认抗纤维化因子,可由巨噬细胞以内分泌、自分泌和旁分泌形式特异性作用于靶细胞,不仅减少巨噬细胞浸润,而且减轻间质纤维化。因此定向调节巨噬细胞分泌的细胞因子种类及数量,也可成为调节炎症和纤维化的方向。
5 总结与展望
巨噬细胞参与肾间质纤维化过程。以巨噬细胞为首的炎症细胞向肾间质募集的确切机制尚不十分清楚。随着研究的进展,巨噬细胞本身及在肾纤维化中的作用有了新发现。针对巨噬细胞的靶向治疗可通过减少肾间质巨噬细胞聚集及抑制其活化来减轻肾脏炎症反应,定向诱导具有抗炎抗纤维化作用的巨噬细胞活化或细胞因子分泌,从而有效阻断肾小管间质纤维化,成为临床治疗肾间质纤维化的新思路和策略。
1 Becker GJ,Hewitson TD.The role of tubulointerstitial injury in chronic renal failure〔J〕.Curr Opin Nephrol Hypertens,2000;9(2):133-8.
2 Filiopoulos V,Vlassopoulos D.Inflammatory syndrome in chronic kidney disease:pathogenesis and influence on outcomes〔J〕.Inflamm Allergy Drug Targets,2009;8(5):369-82.
3 Ferenbach D,Kluth DC,Hughes J.Inflammatory cells in renal injury and repair〔J〕.Semin Nephrol,2007;27(3):250-9.
4 Nikolic-Paterson DJ,Atkins RC.The role ofmacrophages in glomerulonephritis〔J〕.Nephrol Dial Transplant,2001;16(Suppl 5):3-7.
5 Wang Y,Wang YP,Zheng G,et al.Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease〔J〕.Kidney Int,2007;72(3):290-9.
6 Lange-Sperandio B,Cachat F,Thornhill BA,et al.Selectins mediate macrophage infiltration in obstructive nephropathy in newbornmice〔J〕.Kidney Int,2002;61(2):516-24.
7 Cheng QL,Chen XM,Li F,et al.Effects of ICAM-1 antisense oligonucleotide on the tubulointerstitium inmicewith unilateral ureteral obstruction〔J〕.Kidney Int,2000;57(1):183-90.
8 Vielhauer V,Anders HJ,Mack M,et al.Obstructive nephropathy in the mouse:progressive fibrosis correlateswith tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2-and 5-positive leukocytes〔J〕.JAm Soc Nephrol,2001;12(6):1173-87.
9 Wada T,Furuichi K,Sakai N,et al.Gene therapy via blockade ofmonocyte chemoattractant protein-1 for renal fibrosis〔J〕.JAm Soc Nephrol,2004;15(4):940-8.
10 Kitagawa K,Wada T,Furuichi K,et al.Blockade of CCR2 ameliorates progressive fibrosis in kidney〔J〕.Am JPathol,2004;165(1):237-46.
11 Yamagishi H,Yokoo T,Imasawa T,et al.Genetically modified bone marrow-derived vehicle cells site specifically deliver an anti-inflammatory cytokine to inflamed interstitium of obstructive nephropathy〔J〕.JImmunol,2001;166(1):609-16.
12 Metcalfe PD,Leslie JA,Campbell MT,et al.Testosterone exacerbates obstructive renal injury by stimulating TNF-alpha production and increasing proapoptotic and profibrotic signaling〔J〕.Am JPhysiol Endocrinol Metab,2008;294(2):E435-43.
13 Tan X,Wen X,Liu Y.Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-kappaB signaling〔J〕.JAm Soc Nephrol,2008;19(9):1741-52.
14 Guo S,Wietecha TA,Hudkins KL,et al.Macrophages are essential contributors to kidney injury inmurine cryoglobulinemicmembranoproliferative glomerulonephritis〔J〕.Kidney Int,2011;80(9):946-58.
15 Lin SL,Castano AP,Nowlin BT,et al.Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations〔J〕.J Immunol,2009;183(10):6733-43.
16 Lange-Sperandio B,Schimpgen K,Rodenbeck B,et al.Distinct roles of Mac-1 and its counter-receptors in neonatal obstructive nephropathy〔J〕.Kidney Int,2006;69(1):81-8.
17 Persy VP,Verhulst A,Ysebaert DK,etal.Reduced postischemicmacrophage infiltration and interstitial fibrosis in osteopontin knockout mice〔J〕.Kidney Int,2003;63(2):543-53.
18 Sung SA,Jo SK,ChoWY,etal.Reduction of renal fibrosisasa resultof liposome encapsulated clodronate induced macrophage depletion after unilateral ureteral obstruction in rats〔J〕.Nephron Exp Nephrol,2007;105(1):e1-9.
19 Eddy AA.Progression in chronic kidney disease〔J〕.Adv Chronic Kidney Dis,2005;12(4):353-65.
20 Ninichuk V,Khandoga AG,Segerer S,etal.The role of interstitialmacrophages in nephropathy of type 2 diabetic db/db mice〔J〕.Am J Pathol,2007;170(4):1267-76.
21 Henderson NC,Mackinnon AC,Farnworth SL,et al.Galectin-3 expression and secretion linksmacrophages to the promotion of renal fibrosis〔J〕.Am JPathol,2008;172(2):288-98.
22 Pilling D,Tucker NM,Gomer RH.Aggregated IgG inhibits the differentiation of human fibrocytes〔J〕.JLeukoc Biol,2006;79(6):1242-51.
23 Lange-Sperandio B,Trautmann A,Eickelberg O,et al.Leukocytes induce epithelial to mesenchymal transition after unilateral ureteral obstruction in neonatalmice〔J〕.Am JPathol,2007;171(3):861-71.
24 Vielhauer V,KulkarniO,Reichel CA,etal.Targeting the recruitmentof monocytes and macrophages in renal disease〔J〕.Semin Nephrol,2010;30(3):318-33.
25 Nishida M,Okumura Y,Fujimoto S,et al.Adoptive transfer ofmacro-phages ameliorates renal fibrosis in mice〔J〕.Biochem Biophys Res Commun,2005;332(1):11-6.
26 Nishida M,Hamaoka K.Macrophage phenotype and renal fibrosis in obstructive nephropathy〔J〕.Nephron Exp Nephrol,2008;110(1):e31-6.
27 Mosser DM.Themany faces ofmacrophage activation〔J〕.JLeukoc Biol,2003;73(2):209-12.
28 Kushiyama T,Oda T,Yamada M,et al.Alteration in the phenotype of macrophages in the repair of renal interstitial fibrosis in mice〔J〕.Nephrology(Carlton),2011;16(5):522-35.
29 Anders HJ,Ryu M.Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis〔J〕.Kidney Int,2011;80(9):915-25.
30 Duffield JS.The inflammatory macrophage:a story of Jekyll and Hyde〔J〕.Clin Sci(Lond),2003;104(1):27-38.
31 Watkins SK,Egilmez NK,Suttles J,et al.IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo〔J〕.J Immunol,2007;178(3):1357-62.
R692
A
1005-9202(2012)16-3603-04;
10.3969/j.issn.1005-9202.2012.16.133
黑龙江省卫生厅科研基金(2010-099);哈尔滨医科大学附属第二医院青年基金(QN2011-21)
安惠霞(1962-),女,博士,教授,主任医师,主要从事肾脏疾病及肾间质纤维化研究。
陈 晓(1984-),男,硕士,主要从事肾间质纤维化研究。
〔2011-10-19收稿 2012-01-27修回〕
(编辑 袁左鸣/张 慧)
