贵金属修饰的TiO2催化剂湿式氧化处理有机酸废水
2012-01-10陈航宁郑育元郭宗英汪国军吴粮华顾松园
陈航宁,郑育元,郭宗英,汪国军,郭 耘,吴粮华,顾松园
(1.中国石油化工股份有限公司上海石油化工研究院,上海 201208;2.华东理工大学化工学院,上海200237)
贵金属修饰的TiO2催化剂湿式氧化处理有机酸废水
陈航宁1,2,郑育元1,郭宗英1,汪国军1,郭 耘2,吴粮华1,顾松园1
(1.中国石油化工股份有限公司上海石油化工研究院,上海 201208;2.华东理工大学化工学院,上海200237)
以纳米TiO2为载体,采用浸渍法制备了贵金属(Ru,Pd和Pt)质量分数为1%的催化剂,利用X射线衍射(XRD)、N2物理吸附(N2physical adsorption)、程序升温还原(H2-TPR)和透射电镜(TEM)等表征手段对催化剂进行了表征,并利用催化剂进行湿式氧化处理含有机酸的废水。结果表明,添加Ru或Pt可以有效提高TiO2的氧化活性,而Pd对氧化活性提高不明显。在处理丙烯酸废水时,与Pt/TiO2催化剂相比,Ru/TiO2催化剂具有更高的低温氧化活性,在反应温度130 ℃,初始氧气压力3.0 MPa下,丙烯酸废水的化学需氧量(COD)去除率达到91.3%,而在处理饱和有机酸废水(如乙酸和丁二酸)时,Ru/TiO2催化剂的氧化活性高于Pt/TiO2。
湿式氧化 贵金属催化剂 二氧化钛 有机酸废水
在化工生产过程中,作为反应溶剂、介质或冷却液的水不可避免地受到有机物的污染[1],该类污染物具有排放量大、污染面广和生物降解困难等特点。由于利用传统的生物处理技术很难有效地降解该类废水中的有机物,20世纪50年代美国科学家Zimmermann发明了一种处理有毒、有害、高浓度有机废水的湿式氧化法[2,3]。该法是在以空气或纯氧为氧化剂,在液相中将有机污染物氧化为CO2和水等小分子化合物,具有应用范围广,处理效率高,氧化速率快和二次污染少等特点,并且在反应过程中不产生NOx,SO2,HCl和CO等有害气体,此外,设备占地面积小[4]。湿式氧化技术可以单独处理废水,也可以作为生化处理的预处理,通过湿式氧化,降低废水的化学需氧量(COD),去除对生化细菌有毒有害的物质,进而提高废水的可生化性。在湿式氧化处理有机废水的过程中,常生成一些小分子有机酸。这些有机酸很难通过传统的湿式氧化法去除,因此,研究一种湿式氧化催化剂,以提高湿式氧化反应的效率显得尤为重要。贵金属负载型催化剂具有很高的加氢和氧化活性、稳定性和耐腐蚀性等优点,已被广泛用于石油化工和汽车尾气处理[5]。在催化湿式氧化反应中,研究较多的贵金属有Ru,Rh,Pd,Au,Pt和Ir[6]。Gomes等[7]以1%Pt/C为催化剂,采用湿式氧化分别处理乙酸、丙酸和丁酸的模拟废水,发现有机酸的转化率分别达到93%,96%和75%。Gündüz等[8]研究了TiO2负载Ru,Pd和Pt贵金属催化剂在湿式氧化处理丁酸和马来酸废水中的活性。该研究由于采用温和的反应条件(反应温度为 60 ℃,氧气压力为常压),Ru,Pd和Pt催化剂活性均很低,反应3 h后,丁酸和马来酸的转化率均低于2.3%,因此很难比较出何种贵金属催化剂的氧化活性更高。本研究选取纳米TiO2为载体,利用浸渍法制备了Ru,Pd和Pt负载型催化剂,并将其用于湿式氧化处理含丙烯酸、乙酸和丁二酸有机废水;利用N2物理吸附(N2physical adsorption)、X射线衍射(XRD)、透射电镜(TEM)和程序升温还原(H2-TPR)等手段对制备的催化剂进行了表征,为湿式氧化处理工业有机酸废水的多相催化剂开发提供了理论依据。
1 实验部分
1.1 催化剂制备
催化剂均采用浸渍法制备:按照贵金属负载质量分数为1%,将一定浓度的贵金属盐(RuCl3,PdCl2或H4PtCl6)溶液加入纳米TiO2粉末中,将该混合溶液超声处理30 min后,在80 ℃下干燥12 h,然后在400 ℃的空气或氢气气氛中焙烧2 h。氢气还原的催化剂记为Ru/TiO2-H,Pd/TiO2-H和Pt/TiO2-H,空气气氛中焙烧的催化剂记为Ru/TiO2-O,Pd/TiO2-O和Pt/TiO2-O。
1.2 催化剂表征
催化剂的比表面积、孔容和孔径分布在Micromeritics TriStar 3000型多通道物理吸附仪上分析测定,操作温度-196 ℃,比表面积和孔径数据分别根据Brunauer-Emmett-Teller(BET)和Barret-Joyner-Halenda(BJH)模型计算得到。催化剂形貌分析在FEI Tecnai 20 S-TWIN(200KV)仪器上进行。催化剂晶型结构采用德国Bruker D8 Advance型X射线衍射仪分析,Cu靶,Kα射线源,林克斯探测器,管电压40 kV,管电流40 mA,扫描速率为5 (°)/min,2θ为10~90°。H2-TPR表征在Micromoritics公司的Autochemisorption AnalyzerⅡ2920型化学吸附仪上进行,催化剂先在室温下用氦气吹扫10 min,再通入H2和Ar(体积分数分别为10%和90%)的混合气,气体流量50 mL/min,以10 ℃/min的速率由室温升至800 ℃,用TCD检测H2的消耗量。
1.3 催化剂活性评价
催化剂的活性评价在1 L高压反应釜中进行。分别将4 g催化剂,600 mL配制的乙酸、丁二酸、丙烯酸模拟废水加入釜中,室温下充入氧气至3 MPa,当温度升至反应温度时,开启搅拌至400 r/min,2 h后停止搅拌并冷却停止反应。配制的乙酸、丁二酸和丙烯酸模拟废水的COD值分别为6 610,6 290和7 380 mg/L。COD值采用美国Hach DR2800分析仪测定,并根据分析结果计算COD去除率。
2 结果与讨论
2.1 表征结果
催化剂的比表面积、孔容和孔径等物性参数见表1。从表可知,纳米TiO2载体比表面积为182.0 m2/g,负载质量分数为1%的贵金属后,比表面积明显降低,Ru,Pd和Pt分别修饰的TiO2催化剂比表面积都为100.0 m2/g左右,非常相近,并且所有样品的孔容基本保持不变,修饰贵金属之后孔径有所提高。结合透射电镜图(图1)可以看出,纳米TiO2颗粒间有团聚现象,N2物理吸附实验所测的平均孔径为载体团聚后所形成的空隙。由于多相催化湿式氧化是气-固-液三相反应,比表面积较大的催化剂可以有效地促进有机物、氧气和催化剂三者间的接触,进而提高湿式氧化的效率。由于本研究制备的贵金属修饰 TiO2催化剂比表面积、孔容和孔径参数均非常接近,因此,三种催化剂的氧化活性差异受上述参数的影响很小。

表1 催化剂物性参数Table 1 Physical parameters of different catalysts
图1是TiO2上负载质量分数为1%的Ru,Pd和Pt贵金属的透射电镜结果。可以看出,实验所采用的TiO2粉体直径为10 nm左右。另外,贵金属Ru,Pd和Pt以纳米颗粒形式高度分散在TiO2载体上,且无团聚现象。

图1 Ru,Pd和Pt贵金属催化剂的透射电镜分析结果Fig.1 TEM images of Ru/TiO2-H,Pd/TiO2-H and Pt/TiO2-H catalysts
图2为载体TiO2及TiO2负载Ru,Pd和Pt催化剂的XRD谱图。从图中可以看出,实验选用的TiO2载体为典型的锐钛矿结构,利用Scherrer公式计算TiO2平均粒径为9.8 nm。负载贵金属Ru,Pd和Pt并在400 ℃氢气气氛下还原后,TiO2晶体粒径略有增加,分别为11.4,11.9和11.9 nm。Pt/TiO2催化剂的XRD谱图在39.7°处显示有微弱的Pt(100)XRD特征衍射峰[9],而Ru和Pd贵金属催化剂未能在XRD上检测到Ru(38.4°和44.2°)[10]及Pd(40.1°和44.6°)的特征峰[11],表明Ru和Pd纳米颗粒高度分散在TiO2载体上,且分散度高于Pt,该结果与透射电镜结果相符。另外,与TiO2载体相比,Ru,Pd和Pt催化剂在32°附近出现了一个小峰,可能是在催化剂制备过程中,部分TiO2晶型由锐钛矿转变成了板钛矿结构。
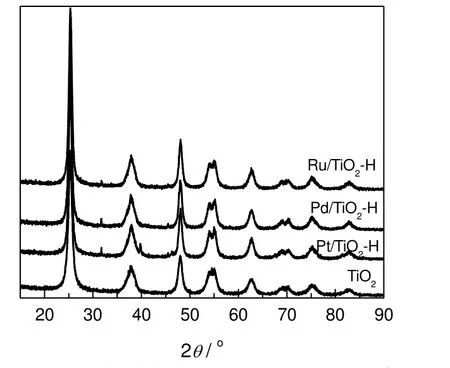
图2 TiO2和负载贵金属的TiO2的XRD图谱Fig. 2 XRD patterns of TiO2 and noble metal supported TiO2 catalysts

图3 TiO2和负载贵金属的TiO2的H2-TPR图谱Fig.3 H2-TPR profiles of TiO2 and noble metal supported TiO2 catalysts
图3为载体TiO2及TiO2负载Ru,Pd和Pt且未经过焙烧或氢气还原的催化剂的H2-TPR谱图。TiO2载体还原峰出现在400 ℃以后,且氢气吸附量很少。与载体相比,负载贵金属Ru,Pd和Pt的TPR谱图发生了较大变化。Pt/TiO2催化剂的第一个还原峰出现在115 ℃,表明在此温度附近,Ptn+还原成Pt0,在200 ℃附近,TPR谱图出现一个负峰,可以解释为H2在较低温度下被Pt0解离成H原子,随后H原子迁移到TiO2载体上,发生了反溢氢现象,释放出了H2[12,13]。Pd/TiO2催化剂第一个微弱的还原峰出现在150 ℃附近,此为颗粒较小且与载体作用较弱的Pdn+的还原峰,237 ℃左右的还原峰则为与TiO2载体作用较强的Pdn+的还原峰[14]。Ru/TiO2的H2-TPR在90和212 ℃附近出现两个还原峰,分别代表高分散的Run+和与载体作用较强的Run+的还原峰[15,16],而在300 ℃附近,负载Ru,Pd和Pt的催化剂均出现了一个较弱的还原峰,该峰属于TiO2载体表面氧的还原峰,证明添加贵金属后,可有效降低Ti-O键能,增加了催化剂晶格氧的流动性,从而促进了催化剂的氧化活性[12,13]。
2.2 催化湿式氧化处理丙烯酸废水
以丙烯酸模拟废水为反应底物,对空气气氛下焙烧或氢气气氛下还原的Pd/TiO2,Pt/TiO2和Ru/TiO2催化剂的氧化活性进行了评价。从表2可以看出,在反应温度200 ℃,初始氧压3 MPa,添加纳米TiO2对丙烯酸废水COD去除率只有46.7%,与未加催化剂相比,湿式氧化效率没有明显提高。以氢气还原制得的Pd/TiO2和Pt/TiO2催化剂氧化活性略高于空气气氛下焙烧的催化剂,而Ru/TiO2经氢气还原后活性则有了较大幅度提高。Ru/TiO2-O催化剂对丙烯酸废水COD去除率只有48.1%,而Ru/TiO2-H催化剂对COD去除率达到94.4%。从表2的结果可知,在处理丙烯酸废水时,氧化活性由大到小为Pt ≈ Ru>Pd。

表2 Pd/TiO2,Pt/TiO2和Ru/TiO2催化剂处理丙烯酸废水活性比较Table 2 The comparison of Pd/TiO2, Pt/TiO2 and Ru/TiO2 on catalytic wet oxidation(CWO) of acrylic acid
为进一步分析Pt/TiO2和Ru/TiO2催化剂在处理丙烯酸废水中的氧化活性,本研究还考察了催化剂在不同反应温度下的氧化活性。从图 4可以看出,Pt/TiO2催化剂在100 ℃下即表现出了氧化活性,反应2 h后COD去除率达到47.9%,而Ru/TiO2催化剂在120 ℃下COD去除率只有31.4%。当反应温度升高至130和160 ℃时,而Pt/TiO2催化剂的COD去除率分别提高到了91.3%和94.1%,继续升高反应温度COD去除率提高较小,而Ru/TiO2催化剂在180℃下COD的去除率才达到94.4%。实验数据表明,在处理丙烯酸废水时Pt/TiO2催化湿式氧化活性高于Ru/TiO2。
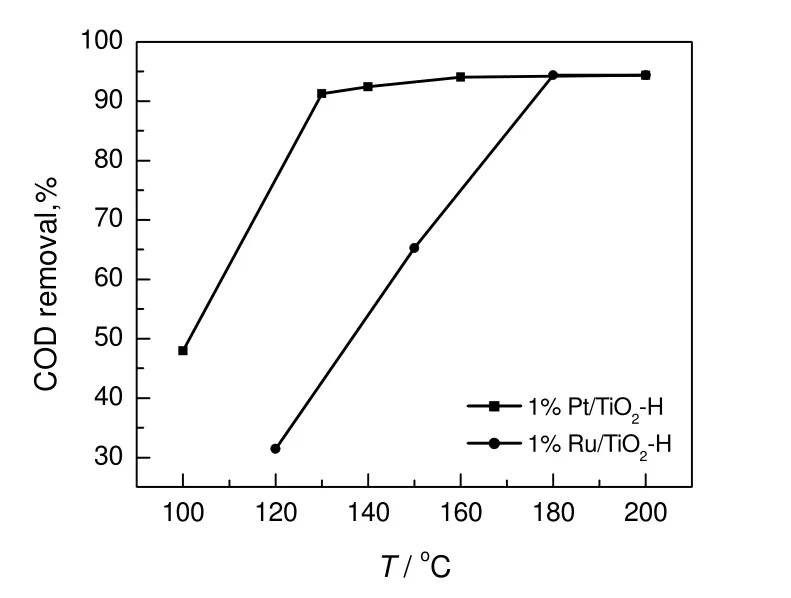
图4 Pt/TiO2和Ru/TiO2在不同反应温度下处理丙烯酸废水Fig.4 CWO of acrylic acid over Pt/TiO2 and Ru/TiO2 at different temperature
2.3 催化湿式氧化处理其他有机废水
为了比较Pt/TiO2和Ru/TiO2催化剂在处理其他有机废水时的氧化活性,本研究选取了乙酸和丁二酸废水为催化湿式氧化的反应底物,在反应温度200 ℃,初始氧压3 MPa的条件下反应,结果见表3。从表3的结果可看出,利用Pt/TiO2催化剂处理乙酸和丁二酸废水时,COD去除率分别只有13.7%和26.1%,而利用Ru/TiO2作为催化剂,COD去除率分别为63.1%和89.7%。因此,在处理此类废水时,Ru的氧化活性高于Pt。

表3 Pt/TiO2和Ru/TiO2催化剂处理各种废水结果Table 3 The results of CWO of different wastewaterover Pt/TiO2 and Ru/TiO2
从N2物理吸附、TEM和XRD表征结果可以看出,Ru,Pd和Pt贵金属在TiO2载体上分散度均较高,三种催化剂比表面积、孔容和孔径参数相近,但是在湿式氧化有机酸时却表现出了不同的催化活性。如Pt/TiO2虽然在处理饱和一元有机酸和二元有机酸时活性较差,但在处理丙烯酸时活性高于 Ru/TiO2催化剂,Pd/TiO2催化活性在三种贵金属中为最低。H2-TPR结果表明,Ru,Pt修饰的TiO2催化剂在115 ℃以下即出现还原吸收峰,表明Run+和Ptn+在TiO2上高度分散且与载体相互作用较弱,而Pdn+需较高温度才能还原为0价,证明其与载体间存在强相互作用,因而Ru和Pt负载的TiO2催化剂氧化活性高于Pd/TiO2。
3 结 论
a)以纳米TiO2为载体制备了Ru,Pd和Pt贵金属催化剂,三种催化剂具有较高的比表面积,贵金属在TiO2载体上分散度高,无团聚。添加Ru,Pd和Pt贵金属可以活化TiO2表面晶格氧,从而提高TiO2的氧化活性。
b)Pt催化剂在处理丙烯酸废水时活性最高,其次是Ru催化剂和Pd催化剂。氢气还原的贵金属催化剂活性高于空气气氛下焙烧的催化剂,其中氢气还原的Ru催化剂催活性提高幅度最大。
c)Ru/TiO2催化剂在乙酸和丁二酸催化湿式氧化反应中的活性高于Pt/TiO2。
[1]Levec J, Pintar A. Catalytic wet-air oxidation processes: a review [J]. Catalysis Today, 2007, 127, 172-184.
[2]Zimmermann F J. Wet air oxidation of hazardous organic in wastewater: 美国, 2665249 [P]. 1950.
[3]Zimmermann F J. New waste disposal process[J]. Chem Eng, 1958, 56: 117.
[4]孙德智, 于秀娟, 冯玉杰. 环境工程中的高级氧化技术[M]. 北京:化学工业出版社, 2002: 85-129.
[5]陈 莎, 张 颖, 曹 莹, 等. 催化湿式氧化法处理有机废水的催化剂研究[J]. 环境科学与管理, 2007,32(3): 119-123.Chen sha, Zhang Ying, Cao Ying, et al. Study on the treatment of catalytic wet air oxidation to organic wastewater[J]. Enviromental Science and Mangement, 2007, 32(3): 119-123.
[6]Kim K H, Ihm S K. Heterogeneous catalytic wet air oxidation of refractory organic pollutants in industrial wastewaters: a review[J]. J Hazard Mater, 2011, 34: 160-186.
[7]Gomes H T, Serp P, Kalck P, et al. Carbon supported platinum catalysts for catalytic wet air oxidation of refractory carboxylic acids[J]. Top Catal, 2005, 33: 59-68.
[8]Dukkancı M, Gündüz G. Catalytic wet air oxidation of butyric acid and maleic acid solutions over noble metal catalysts prepared on TiO2[J].Catal Commun, 2009, 10: 913-919.
[9]Mu Y, Liang H, Hu J, et al. Controllable Pt nanoparticle deposition on carbon nanotubes as an anode catalyst for direct methanol fuel cells[J]. J Phys Chem B, 2005, 109: 22212-22216.
[10]Chen C-W, Chen C-Y, Huang Y-H. Method of preparing Ru-immobilized polymer-supported catalyst for hydrogen generation from NaBH4solution[J]. International Journal of Hydrogen Energy, 2009, 34: 2164-2173.
[11]Chin Y H, Dagle R, Hu J, et al. Steam reforming of methanol over highly active Pd/ZnO catalyst[J]. Catalysis Today, 2002, 77: 79-88.
[12]何运兵, 纪红兵. TiO2纳米管负载铂催化剂催化完全氧化甲醛[J]. 材料研究与应用, 2010, 4(4): 387-401.
He Yunbing, Ji Hongbing. Complete catalytic oxidation of formaldehyde over Pt-supported TiO2nanotubes[J]. Materials Research and Application, 2010, 4(4): 387-401
[13]Zhang C, He H, Tanaka K. Catalytic performance and mechanism of a Pt/TiO2catalyst for the oxidation of formaldehyde at room temperature[J].Appl Catal B: Environ, 2006, 65: 37-43.
[14]Shen W J, Okumura M, Matsumura Y, et al. The influence of the support on the activity and selectivity of Pd in CO hydrogenation[J]. Appl Catal A: General, 2001, 213: 225-232.
[15]Basińska A, Klimkiewicz R, Domka F. Ru/Fe2O3catalysts inn-butanol conversion[J]. Appl Catal A: General, 2001, 207: 287-294.
[16]王建强. 新型钌催化剂的制备表征及苯选择加氢反应研究[D]. 上海: 复旦大学化学系, 2005.
Wet Oxidation of Wastewater Containing Organic Acids over TiO2Supported Noble Metal Catalyst
Chen Hangning1,2, Zheng Yuyuan1, Guo Zongying1, Wan Guojun1, Guo Yun2, Wu Lianghua1, Gu Songyuan1
(1. SINOPEC Shanghai Research Institute of Petrochemical Technology, Shanghai 201208, China;2.School of Chemical Engineering,East China University of Science and Technology,Shanghai 200237, China)
Catalysts for catalytic wet oxidation (CWO) of wastewater contaminated by organic acids were prepared by impregnation of noble metals Ru, Pd and Pt on nano TiO2support. These catalysts were characterized by X-ray diffraction (XRD), N2physical adsorption, H2-temperature programmed reduction(H2-TPR) and transmission electron microscope(TEM). The experimental data showed that TiO2oxidation activity was significantly enhanced by adding 1% (mass fraction) Ru or Pt, while it was less effective by adding Pd. In the CWO of acrylic acid, Pt/TiO2catalyst showed much higher oxidation activity than Ru/TiO2under a mild temperature condition. Chemical oxygen demand (COD) removal reached 91.3% under the conditions of temperature 130 ℃ and O2initial pressure 3.0 MPa over Pt/TiO2catalyst. However, Ru/TiO2catalyst had higher activity than Pt/TiO2in the CWO of saturated organic acids, such as acetic acid and succine acid. Therefore, the component of CWO catalyst should be changed when treating wastewater with different kinds of organic acids.
catalytic wet oxidation; noble metal catalyst; titania; wastewater with organic acids
TQ323.8 文献标识码:A
1001—7631 ( 2012 ) 04—0325—05
2012-05-28;
2012-08-14
陈航宁(1982—),男,博士研究生;顾松园(1964—),男,教授级高工,通讯联系人。E-mail: gusy.sshy@sinopec.com
