核磁共振法测定西洛他唑含量
2012-01-08杨金霞姚婷玉王健伟张丹
杨金霞,姚婷玉,王健伟,张丹
(苏州高新技术创业服务中心生物医药分析测试中心,江苏苏州 215011)
核磁共振法测定西洛他唑含量
杨金霞,姚婷玉,王健伟,张丹
(苏州高新技术创业服务中心生物医药分析测试中心,江苏苏州 215011)
建立核磁共振(1H NMR)法测定西洛他唑含量的方法。采用Bruker Avance Ⅲ400 MHz核磁共振波谱仪,以1,2,4,5-四氯-3-硝基苯为内标,氘代氯仿为溶剂,zg30脉冲序列。对实验参数如扫描次数、延迟时间等进行优化。西洛他唑质量在5~30 mg范围内,样品和内标物的积分面积比值与样品和内标物的质量比值呈现良好的线性关系,线性相关系数r=0.995 3,测定结果的相对标准偏差小于0.4%(n=5)。该方法可用于药品的质量控制和监管。
核磁共振法;西洛他唑;含量
西洛他唑(cilostazol)临床上主要用于改善慢性动脉闭塞症引起的缺血性症状,如溃疡、肢痛、发冷及间歇性跛行[1]。经查阅相关文献及国家药品标准[2,3],目前测定西洛他唑的方法有高效液相色谱法(HPLC),该法以甲醇-水为流动相,定量检测需要对照品,且检测时间较长。中国药典2010年二部附录Ⅸ[4]首次引入核磁共振波谱法,但没有具体品种药物的NMR定量方法。随着现代仪器灵敏度的不断提高,NMR定量法测定药物含量将成为今后发展的趋势[5,6]。
笔者采用1H NMR法测定西洛他唑含量,通过对西洛他唑结构(图1)分析,选择西洛他唑结构中5,7,8三个氢的吸收为特征峰,用已知纯度的标准品1,2,4,5-四氯-3-硝基苯中1个氢为定量内标,根据核磁信号强度与质子数目成正比的特点对药物进行定量分析[4]。该法简便、专属性较强,不需要西洛他唑对照品,对样品无破坏,准确性、重复性较好。
1 实验部分
1.1 主要仪器与试剂
核磁共振波谱仪:Bruker Avance Ⅲ400 MHz型,瑞士Bruker公司;
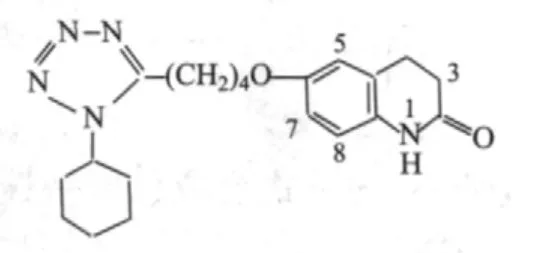
图1 西洛他唑化学结构式
BBO探头:5 mm;
核磁管:内径5 mm;
电子天平:XS205,d=0.01 mg,瑞士梅特勒-托利多公司;
溶剂:CDCl3,氘代度 99.8%+0.03%TMS,美国CIL公司;
定量内标:1,2,4,5-四氯-3-硝基苯,批号421-7A,纯度99.3%,美国Chem Service 公司;
西洛他唑原料:苏州中化药品工业有限公司。
1.2 实验条件
采用zg30脉冲序列[7];测定温度(探头温度):300 K;溶剂:CDCl3;采样次数(NS):8,TD=65 536;谱宽:8 012.82 Hz;脉冲宽度(P1):10.60 μs;脉冲功率(PL1):-2.00 dB;脉冲延迟时间(d1):15 s;采样时间(AQ):4.0 894 966 s。
1.3 实验方法
精密称取西洛他唑30 mg和1,2,4,5-四氯-3-硝基苯10 mg置于直径5 mm的核磁管中,加入1.0 mL CDCl3使其完全溶解,盖上管帽,按1.2实验条件进行氢谱检测,调节仪器参数,匀场,采样,图谱处理,确定结构无误后,选择西洛他唑的特征峰(7.6≤δ≤ 7.8)和 1,2,4,5-四氯-3-硝基苯的单峰(6.6≤δ≤6.8)作为定量分析的特征峰,将其积分,取5次测定结果的平均值,按1.4中公式计算西洛他唑的含量。
1.4 结果计算
西洛他唑含量按下式计算:
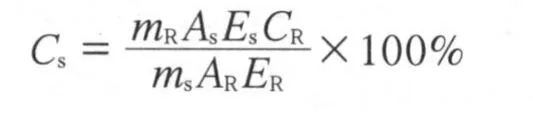
式中:CS——待测样品的含量;
mR——内标物的质量,mg;
AS,AR——样品和内标物的定量特征峰积分面积;
ES,ER——样品和内标物的定量特征峰质子当量质量(以分子量除以定量特征峰对应的质子数),Da;
CR——内标物的含量;
ms——样品质量,mg。
2 结果与讨论
2.1 溶剂的选择
当选择CDCl3为溶剂时,样品和内标物能够完全溶解,且溶剂峰(δ=7.72)与样品的定量特征峰(δ=6.70)完全分离,不相互干扰,测定结果准确,因此选择CDCl3为溶剂。
2.2 内标物的选择
试验发现,1,2,4,5-四氯-3-硝基苯只有一个特征参考峰(δ=7.74)与样品西洛他唑的特征峰(δ=6.70)完全分离,而且这两组峰与其它峰也能完全分开,峰两侧的基线平坦,其积分值比较准确,可作为定量分析的依据。因此选择1,2,4,5-四氯-3-硝基苯为定量内标物。
2.3 样品量的选择
配制不同浓度的样品进行分析,试验结果显示,样品最低浓度为0.01 mol/L即可满足检测要求。
2.4 延迟时间d1的选择
当d1≥5×T1(T1为纵向弛豫时间)才能保证在下次扫描时磁化矢量完全回到Z轴,否则对结果的影响较大。分别设置 d1=1,2,4,6,8,10,15,20,25 s ,进行试验。结果发现,d1≤15 s时,随着d1的增加,样品和内标的峰面积比值逐渐增大;d1>15 s时,样品和内标物的峰面积比值没有明显变化,因此d1选择15 s。
2.5 扫描次数NS的选择
将扫描次数 NS 设定为 4,8,16,32,64,128,对样品分别进行试验,结果表明,扫描次数改变,样品和内标物积分面积的比值没有明显变化,因此选择NS=8。
2.6 工作曲线
精密称取内标物24.8 mg ,置于5 mL容量瓶中,加适量CDCl3充分振摇使其完全溶解,用溶剂CDCl3定容至刻度,作为内标溶液;精密称取西洛他唑 5.4,10.4,15.2,20.1,25.3,30.0 mg 分别加入到核磁管中,各加入内标溶液1.0 mL,使其完全溶解后,按1.2实验条件进行氢谱检测,对样品和内标物的特征峰分别积分,每个浓度样品的积分面积取3次测定结果的平均值,结果见表1。
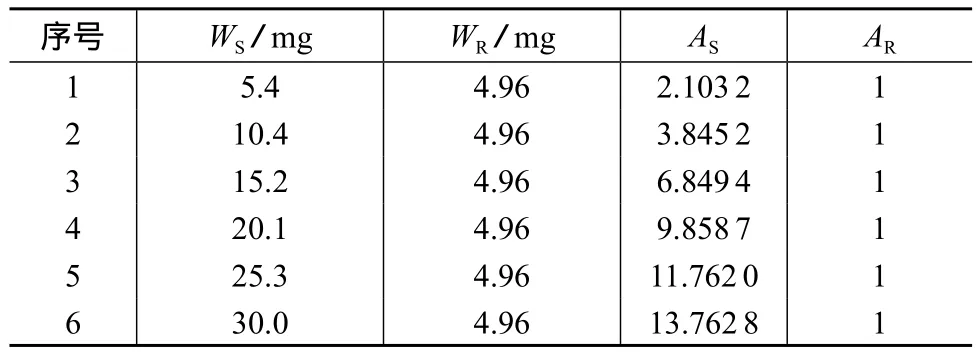
表1 样品与内标物的质量及其特征峰面积
利用样品和内标物积分面积的比值(y)对样品和内标物质量的比值(x)进行线性回归,回归方程为y=2.406 1x-0.523 6,相关系数r=0.995 9,线性范围为 5.0~30.0 mg。
2.7 重复性试验
按1.2实验条件对批号为X21-002,X21-003样品溶液(编号为1#,2#)进行测定,每个样品重复测定5次,计算样品和内标物的特征峰面积的比值,测定结果的相对标准偏差分别为0.12%,0.31%,由此可见方法的重复性较好。
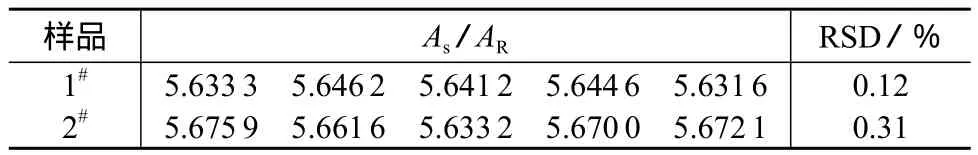
表2 重复性试验结果
2.8 稳定性试验
将批号为X21-003的样品溶液在室温下分别放置 0,2,4,6,8,10,12,24 h 后进行测定,计算样品和内标物特征峰面积的比值,8次测定结果的相对标准偏差为0.36%,表明样品在24 h内放置稳定。
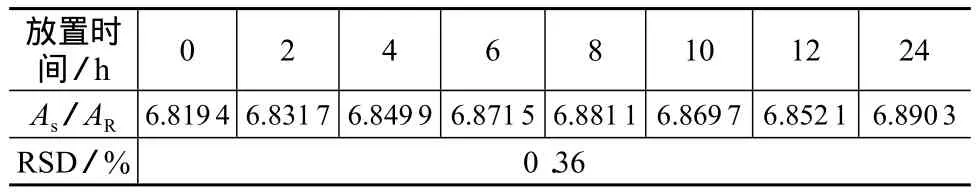
表3 稳定性试验结果
2.9 样品测定
取3批西洛他唑样品分别用本法和标准方法(HPLC法)进行检测,测定结果见表4。由表4可知,核磁共振法与标准方法的测定结果相符合。以1#样品为例,样品、内标物及溶剂的1H NMR谱图见图2。
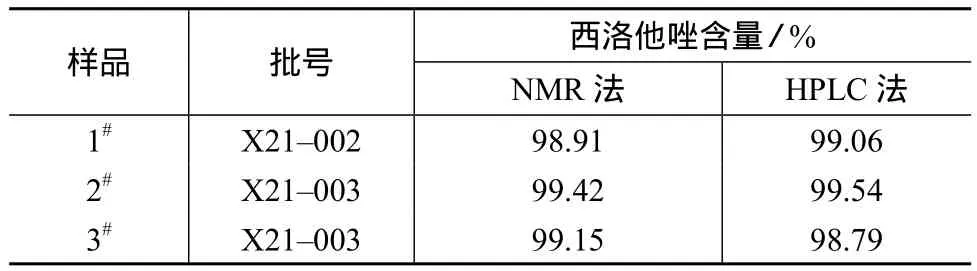
表4 样品测定结果
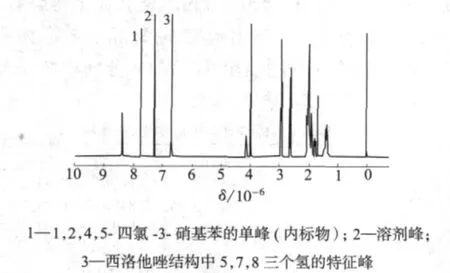
图2 样品、内标物及溶剂的1H NMR谱图
2.10 其它影响因素
(1)为保证NMR定量结果的重现性和准确性,图谱的处理很关键:首先要保证匀场要好,否则会造成半峰宽的加宽和峰型的变化,影响积分面积。匀场好坏的判断标准为:氘代试剂中的内标物TMS除了出现尖锐的单峰外,在其峰的底部两侧各有一个小峰。相位调整要仔细,应将谱图放大后调整峰形左、右轮廓线与水平的基线刚好重合,积分面积的起、止点每次要一致,基线要平直,并对该组峰面积的积分取5次测定结果的平均值,以减少误差。
(2)为保证结果的准确性可以再选择样品的另一组信号峰以同样的方法进行定量,然后以这两组峰测量结果的平均值作为含量测定的结果,这样更接近药品含量的真实结果。
3 结语
采用NMR法测定西洛他唑药品含量,测定结果的重复性及稳定性均较好,该方法具有快速、准确、简单的特点。在被测样品没有高纯度对照品或难以得到对照品的情况下,可以作为药品含量测定的补充,在含量检测的同时,对其结构进行确证(即药物鉴定),这对防止假冒、伪劣药品的出现具有极其重要的意义。
[1]Kambayashi J,Liu Y G,Sun B,et al. Ciloslazol as a unique antithrombotic agent [J]. Curr Pharm Des 2003,9(28):2 281-2 291.
[2]王存芳,严启新,李勇.HPLC法测定西洛他唑含量[J].中华医学研究杂志,2006(1): 21-22.
[3]YBH 05382004 西洛他唑[S].
[4]国家药典委员会.中国药典[M].二部:附录.北京:中国医药科技出版社,2010.
[5]孙静霞,张正行.NMR在药物定量分析中的应用[J].药物分析杂志,2005,25(1): 117-122.
[6]于小波,沈文斌,相秉仁.定量核磁共振技术及其在药学领域的应用进展[J].药学进展,2010,34(1): 17-24.
[7]张友杰,刘小鹏.药物核磁共振定量分析参数研究[J].波谱学杂志,2007,24(3): 289-295.
Determination of Cilostazol by1H NMR
Yang Jinxia, Yao Tingyu, Wang Jianwei, Zhang Dan
(Analytical and Testing Center of Bio-Medicine in Suzhou New & High-Tech Innovation Service Center,Suzhou 215011, China )
A method for the determination of cilostazol by1H NMR was established. Proton nuclearmagnetic resonance spectroscopy(1H NMR) spectra were obtained in CDCl3with the internal standard 1,2,4,5-tetr-achloro-3-nitrobenzene and zg30 pulse sequence by using Bruker AvanceⅢ400 MHz spectrometer. Testing parameters such as scanning number,delay time were optimized. Area ratio of sample and internal standard was linear with mass ratio of sample and internal standard in the range of 5.0-30 mg for cilostazol concentration,r=0.995 3. The relative standard deviation of detection results was less than 0.4%(n=5). This method is suitable for quality control and supervision.
NMR; cilostazol; content
O 657.2
A
1008-6145(2012)04-0068-03
10.3969/j.issn.1008-6145.2012.04.021
联系人:杨金霞;E-mail: yjx@csibi.cn
2012-04-17
