HPLC法测定西洛他唑分散片有关物质
2016-09-02任萃文
任萃文
(哈尔滨商业大学生命科学与环境科学研究中心,哈尔滨150076)
HPLC法测定西洛他唑分散片有关物质
任萃文
(哈尔滨商业大学生命科学与环境科学研究中心,哈尔滨150076)
建立西洛他唑分散片有关物质的检测方法.采用C8柱(250 mm×4.6 mm,5μm);流动相A为乙腈-水(25∶75),流动相B为乙腈-水(50∶50);梯度洗脱:0-6 min-10 min-40 min,流动相A∶B的比例为100∶0-50∶50-0∶100-0∶100;检测波长254 nm.结果表明,西洛他唑与各已知杂质及降解产物均良好分离,最低检测限与最低定量限分别为0.1 ng与0.2 ng,精密度良好.本方法操作简单,对西洛他唑分散片有关物质检测快速,准确,专属性强,灵敏度高.
西洛他唑;有关物质;HPLC;梯度洗脱
西洛他唑为抗血小板药物,主要通过抑制磷酸二酯酶活性而用于血栓性疾病的治疗;尤其对肢体动脉缺血性疾病作用显著,是治疗间歇性跛行的首选药物[1-2].目前,国内上市的西洛他唑制剂主要为普通片剂和胶囊,为了使吸收更迅速并给服药困难者提供方便,研制了西洛他唑分散片,本文对所研制的西洛他唑分散片有关物质检测方法进行研究.
文献报道,西洛他唑一般采用高效液相色谱法进行质量控制[3-6],USP35收载了西洛他唑原料的有关物质检测方法,并列出3种可能存在的已知杂质,从其结构(见图1)分析,西洛他唑杂质B与西洛他唑结构相对接近,西洛他唑杂质A和C的结构相差较大,采用一般的等强度洗脱难以实现各已知杂质及主峰的同时分离,故本文选用梯度洗脱方式对本品有关物质检查,并进行方法学研究.

图1 西洛他唑及其杂质结构图
1 仪器与试药
1.1仪器
JASCO高效液相色谱仪(日本),PU-2089泵,UV-2075可见-紫外检测器,AS-2055自动进样器,ChromPass色谱工作站.
1.2试药
西洛他唑对照品(中国食品药品检定研究院,批号:100363-201202);Cilostazol Related Compound A(西洛他唑杂质 A对照品,USP,批号:F0G160);Cilostazol Related Compound B(西洛他唑杂质B对照品,USP,批号:F0G073);Cilostazol Related Compound C(西洛他唑杂质C对照品,USP,批号:F0G074,质量分数99.5%);西洛他唑分散片(自制,批号:140901、140902、140903);乙腈为色谱纯,其他试剂均为分析纯.
2 试验内容与结果
2.1色谱条件
色谱柱:采用phenomenex C8柱(250 mm×4.6 mm,5μm);流动相A为乙腈-水(25∶75),流动相B为乙腈-水(50∶50);梯度洗脱:0-6 min-10 min-40 min,流动相A∶B的比例为100∶0-50 ∶50-0∶100-0∶100;检测波长254 nm;流速:1.0 mL/min;进样量:20μL;理论板数按西洛他唑峰计算不低于2 000.
2.2供试品溶液的配制
精密称取西洛他唑分散片样品细粉适量(约含西洛他唑40 mg),置100 mL量瓶中,加入乙腈40 mL,超声,使西洛他唑溶解后,用蒸馏水定容至刻度,摇匀,滤过,取续滤液作为供试品溶液.
2.3系统适用性试验
取西洛他唑对照品及各杂质对照品适量,加乙腈溶解制成混合溶液.在2.1色谱条件下进样,记录色谱图.西洛他唑及其杂质A、B和C保留时间分别为17.28、4.21、15.52、33.86 min,分离度为3.53、西洛他唑主峰理论塔板数为30 274,西洛他唑与各杂质均能有效分离,见图2.
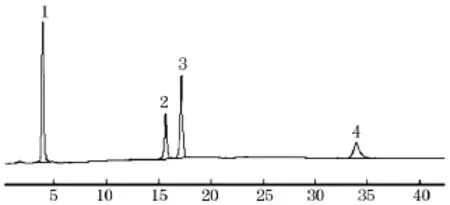
图2 系统适用性图谱
2.4辅料干扰试验
取处方量的西洛他唑分散片空白辅料适量,按
2.2项的方法配制,进样.结果见图3.空白辅料在此波长处没有吸收,不干扰西洛他唑及其杂质的测定.

图3 辅料干扰图谱
2.5专属性试验
酸破坏试验精密称取西洛他唑分散片细粉适量(约含西洛他唑40 mg),置100 mL锥形瓶中,加入2 mol/L的盐酸溶液5 mL,在100℃条件下放置5 h,取出,加2 mol/L的氢氧化钠溶液5 mL中和,加入乙腈40 mL,超声,使西洛他唑溶解后,加入蒸馏水50 mL,摇匀,滤过,取续滤液作为酸破坏溶液.
碱破坏试验精密称取西洛他唑分散片细粉适量(约含西洛他唑40 mg),置100 mL锥形瓶中,加入2 mol/L的氢氧化钠溶液5 mL,在100℃条件下放置5 h,取出,加2 mol/L的盐酸溶液5 mL中和,加入乙腈40 mL,超声,使西洛他唑溶解后,加入蒸馏水50 mL,摇匀,滤过,取续滤液作为碱破坏溶液.
热破坏试验取西洛他唑分散片置120℃烘箱中高温破坏5 h,取出.精密称取细粉适量(约含西洛他唑40 mg),置100 mL量瓶中,加入乙腈40 mL,超声,使西洛他唑溶解后,用蒸馏水定容至刻度,摇匀,滤过,取续滤液作为热破坏溶液.
氧破坏试验精密称取西洛他唑分散片细粉适量(约含西洛他唑40 mg),置100 mL锥形瓶中,加入3%的过氧化氢溶液5 mL,在100℃条件下放置2 h,取出,加入乙腈40 mL,超声,使西洛他唑溶解后,加入蒸馏水45 mL,摇匀,滤过,取续滤液作为氧破坏溶液.
光破坏试验精密称取西洛他唑分散片样品细粉适量(约含西洛他唑40 mg),置100 mL量瓶中,加入乙腈40 mL,超声,使西洛他唑溶解后,用蒸馏水定容至刻度,摇匀,置4 500 Lx下强光照射72 h,取出,滤过,取续滤液作为光破坏溶液.
精密称取西洛他唑分散片样品细粉适量(约含西洛他唑40 mg),置100 mL量瓶中,加入乙腈40 mL,超声,使西洛他唑溶解后,用蒸馏水定容至刻度,摇匀,滤过,取续滤液作为未破坏的供试品溶液;精密吸取上述供试品溶液各20μL,注入色谱仪,记录色谱图.结果表明西洛他唑各降解产物均能和主峰达到良好分离,说明上述色谱系统能有效检测杂质,专属性强.
2.6供试品溶液稳定性试验
取批号为140901的西洛他唑分散片样品,照2.2项供试品溶液的配制方法配制成每1 mL约含西洛他唑0.4 mg的溶液,分别于0、1、2、3、4、5 h测定,记录色谱图,计算峰面积的RSD值,结果见表1.结果表明,本品溶液稳定性较好.
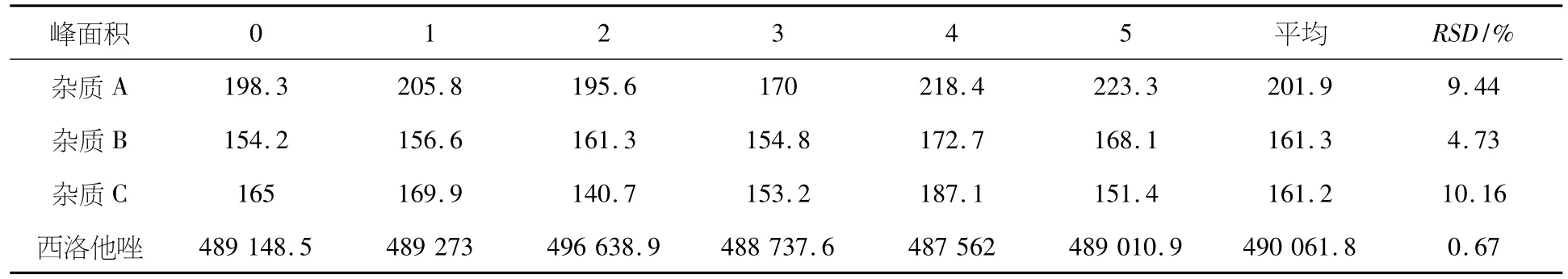
表1 稳定性试验结果
2.7进样精密度试验
精密量取西洛他唑对照品溶液20μL进样测定,连续平行分析6次,记录色谱图,计算主峰面积RSD值为0.71%.
2.8检测限和定量限
将西洛他唑对照品用40%乙腈逐步稀释,进样,记录S/N约为3时的量为最低检测限,S/N约为10时的量为最低定量限,结果本品的检测限为0.1 ng,定量限0.2 ng.
2.9有关物质检测
取西洛他唑分散片样品适量,照2.2供试品溶液的配制方法配制成每1 mL约含西洛他唑0.4 mg的溶液,作为供试品溶液;精密量取1 mL,置100 mL量瓶,用40%乙腈稀释至刻度,摇匀,作为对照溶液.照2.1项下的色谱条件,精密量取对照溶液20μL注入液相色谱仪,调节检测灵敏度,使主成份色谱峰的峰高为满量程的20%,再精密量取供试品溶液和对照溶液各20μL注入液相色谱仪,记录色谱图,结果见表2.
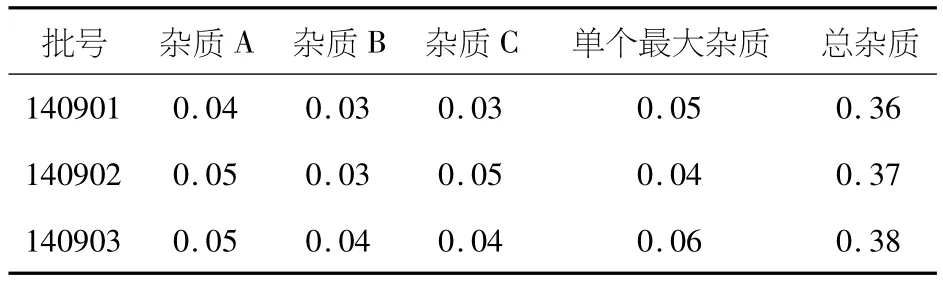
表2 西洛他唑分散片有关物质测定结果(%)
3 讨论
3.1波长的选择
分别取适量浓度的西洛他唑及其杂质对照品溶液在190~400 nm波长范围内进行扫描,结果西洛他唑在254 nm处有最大吸收,西洛他唑杂质A、B和C分别在256、262、259 nm处有最大吸收,综合考虑选择本品的检测波长为254 nm.
3.2色谱柱选择
由于西洛他唑及其已知杂质之间结构的差异,采用极性较弱的C18色谱柱检测本品,西洛他唑杂质A与溶剂峰不能有效分离,还会使检测时间延长;本文采用中等极性的C8色谱柱检测,既能保证西洛他唑杂质A与溶剂峰的效分离,还能缩短检测时间,提高检测灵敏度.经上述方法验证,本品色谱条件重现性好,精准度高.
[1]陈小勇,彭润涛,江宇,等.抗血小板药物西洛他唑市场分析[J].中国医药情报,2003,9(6):39-43.
[2]万文辉,钱晓明.经典抗血小板治疗药物临床研究进展[J].中国误诊学杂志,2011,11(7):1533-1535.
[3]唐坤.反相高效液相色谱法测定西洛他唑片含量[J].华夏医学,2010(3):297-299.
[4]杨金霞.超高效液相色谱法测定西洛他唑含量[J].中国中医药咨讯,2011,3(10):115。
[5]李晓红.HPLC法测定西洛他唑片的有关物质[J].中国药事,2010(12):1219-1220,1247.
[6]郑一美.反相高效液相色谱法检测西洛他唑中有关物质[J].中国药业,2006,15(14):20-21.
Determ ination of cilostazol dispersible tablets related substances by HPLC
REN Cui-wen
(Research Center on Life Sciences and Environmental Sciences,Harbin University of Commerce,Harbin 150076,China)
To establish amethod for cilostazol dispersible tablets relativematerials,the determinationswere carried out on a C8column(250 mm×4.6 mm,5μm)with acetonitrile:water(flow phase A)25∶75-acetonitrile∶water(flow phase B)50∶50 asmobile phase;gradient elution:0min-6min-10min-40min,the ratio of phase A∶phase B was100 ∶0-50∶50-0∶100-0∶100.The detection UV wavelength was 254 nm.The results indicated that the developed method could be used for the determination of cilostazol impurities and itsmetabilite with a good resolution.The limit of detection and limit of quantification of cilostazolwere 0.1 ng and 0.2 ng,respectively.Themethod was proved to be simple,accurate,reproducible and feasible,and can be used for the quality control of cilostazol dispersible tablets.
cilostazol;related substances;HPLC;gradient elution
R927
A
1672-0946(2016)02-0157-04
2015-08-28.
任萃文(1980-),女,硕士,高级工程师,研究方向:制备工艺及质量标准.
