利用Mitsunobu反应合成川芎嗪衍生物*
2011-11-26王慧颖孙平华陈卫民
王慧颖, 孙平华, 陈卫民
(暨南大学 药学院,广东 广州 510632)
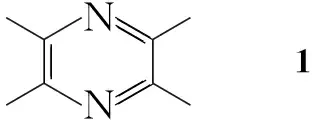
川芎嗪(1)是从川芎中分离出的生物碱单体,具有抑制血小板聚集、抗血栓形成、抗自由基等作用。临床上广泛用于治疗缺血性心、脑血管疾病,冠心病及呼吸系统疾病[1,2]。研究发现,1的吡嗪环是决定其药理作用的重要结构部分,即药效基团,而1的甲基很容易被氧化代谢,生成极性和水溶性都较大的代谢产物迅速排出体外,存在代谢快、半衰期短、生物利用度低等缺点[2~4]。因此,现在多以1为先导化合物进行结构修饰,以阻滞或延缓其甲基的氧化代谢,从而延长作用时间、相对增加药效。
对于1的修饰主要有以下方法:利用H2O2氧化1得到对应的单氮氧化物;再与乙酸酐发生重排反应生成2-乙酰氧基甲基-3,5,6-三甲基吡嗪(M1); M1在碱性条件下水解制得2-羟甲基-3,5,6-三甲基吡嗪(2);继而进行进一步的结构修饰[5~8]。或用高锰酸钾直接氧化1制得3,5,6-三甲基-2-吡嗪羧酸[9]。或以1为原料,NBS(N-溴代丁二酰亚胺)为溴化剂,四氯化碳作溶剂,在过氧化二苯甲酰催化下反应制得关键中间体2-溴甲基-3,5,6-三甲基吡嗪(M2);再对M2作进一步的结构修饰[8]。赵永海等[10]以1为起始原料,经溴化、O-烷基化反应合成了4-[(3,5,6-三甲基吡嗪-2-基)甲氧基]苯甲醛(4a)。
本文改进文献[10]方法,以1为起始原料,经30%H2O2氧化制备2(避开刺激性的中间体M2);2与羟基苯甲醛(3a~3c)经Mitsunobu反应合成了三个3,5,6-三甲基吡嗪-2-基-甲氧基苯甲醛衍生物(4a~4c, Scheme 1),收率87%~92%,其结构经1H NMR,13C NMR和ESI-MS表征。


Scheme1
1 实验部分
1.1 仪器与试剂
Bruker AV300 MHz/400 MHz型核磁共振仪(CDCl3作溶剂,TMS作内标);ABI 4000 Q TRAP型质谱仪(ESI源)。
1,工业品,纯度>99%,上海邦成化工有限公司;偶氮二甲酸二乙酯(DEAD); 其余所用试剂均为市售分析纯,其中DMF, THF和四氯化碳经除水处理。
1.2 合成
(1) 2的合成
在三颈瓶中加入1 6.8 g(50 mmol),冰醋酸12.5 mL和30%H2O26 mL,搅拌下于70 ℃反应4 h;再加30%H2O26 mL,回流反应4 h(TLC跟踪)。冷却至室温,用25%NaOH溶液调至pH 10, CHCl3(3×20 mL)萃取, 合并萃取液,用无水Na2SO4干燥,蒸除CHCl3,加入醋酸酐15 mL,回流反应3 h(TLC跟踪)。减压蒸除过量醋酸酐得黑色浆状物,冷却后用25%NaOH溶液调至pH 12,乙酸乙酯(3×20 mL)萃取,合并萃取液,无水Na2SO4干燥,减压蒸除溶剂后经硅胶柱层析[洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=6 ∶1]纯化得淡黄色晶体2 5.563 g,产率73.2%, m.p.88 ℃~89 ℃;1H NMRδ: 2.40(s, 3H, CH3), 2.52(s, 6H, CH3), 4.29(t,J=4.5 Hz, 1H, OH), 4.68(d,J=4.5 Hz, 2H, CH2); ESI-MSm/z: 153.2{[M+H]+} 。
(2) 用Mitsunobu反应合成4(以4a为例)
在三颈瓶中依次加入2 1.52 g(10 mmol),对羟基苯甲醛(3a) 1.46 g(12 mmol),三苯基磷3.28 g(12.5 mmol)和THF 20 mL, N2保护,搅拌下缓慢滴加偶氮二甲酸二乙酯(DEAD) 2.194 g(12.6 mmol, 2mL),滴毕,于室温反应24 h(TLC跟踪)。减压蒸除溶剂,残渣用乙醚溶解,过滤,滤液用饱和NaHCO3溶液洗涤,无水MgSO4干燥,减压蒸除溶剂后经硅胶柱层析(洗脱剂:A=8 ∶1)纯化得淡黄色固体4a。
用类似方法合成白色固体4b和4c。
4a: 收率92.5%, m.p.75 ℃~ 76 ℃;1H NMRδ: 2.50(s, 6H, CH3), 2.57(s, 3H, CH3), 5.21(s, 2H, OCH2), 7.10(d,J=8.64 Hz, 2H, ArH), 7.81(d,J=8.72 Hz, 2H, ArH), 9.86(s, 1H, CHO);13C NMRδ: 190.28, 163.09, 151.26, 149.50, 148.34, 144.35, 131.50, 129.87, 114. 73, 69.66, 21.23, 20.91, 20.12; ESI-MSm/z: 257.3{[M+H]+} 。
4b: 收率87.5%, m.p.121 ℃~122 ℃;1H NMRδ: 2.49(s, 3H, CH3), 2.51(s, 3H, CH3), 2.59(s, 3H, CH3), 3.78(s, 3H, OCH3), 5.36(s, 2H, OCH2), 7.19(d,J=8.52 Hz, 1H, ArH), 7.40(m, 2H, ArH), 9.75(s, 1H, CHO); ESI-MSm/z: 287.2{[M+H]+} 。
4c: 收率89.7%, m.p.92 ℃~93 ℃;1H NMRδ: 2.48(s, 6H, CH3), 2.55 (s, 3H, CH3), 5.17(s, 2H, OCH2), 7.23(d,J=8.2 Hz, 1H, ArH), 7.38~7.45(m, 2H, ArH), 7.50(t,J=8.2 Hz, 1H, ArH), 9.92(s, 1H, CHO);13C NMRδ: 193.48, 160.68, 153.07, 151.44, 150.31, 146.62, 139.34, 131.65, 125.19, 123.49, 115.15, 71.53, 23.19, 22.88, 22.08; ESI-MSm/z: 257.3{[M+H]+} 。
2 结果与讨论
本实验曾参照文献[10]方法,先合成中间体M2(刺激性过大,同时伴有多溴代物生成,产率较低);再与3a于85 ℃反应8 h反应合成4a,收率83.5%(82.9%[10]),总收率46.6%(以1计算,下同)。
另外以DDC(二环己基碳二亚胺)为催化剂[11],2与3a于130 ℃反应24 h合成4a,收率66.3%,总收率48.5%。该方法存在反应后生成的二环己基脲较难去除干净、反应温度过高、副产物较多、产率较低等缺点。
与文献[10]方法和DCC催化法[11]相比,Mitsunobu反应法条件温和,室温即可进行,操作简便,且产率较高(总收率67.7%)。
[1] 魏丽萍,宋艳春. 川芎嗪临床应用进展[J].中医药信息,2000,(6):20-22.
[2] 杨杰,段玉峰,韩果萍,等. 川芎嗪的结构修饰及生物活性[J].化学通报,2003,66(7):454-458.
[3] 徐睿,李源,黄熙. 川芎嗪药物代谢动力学研究进展[J].安徽中医学院学报,2002,21(1):58-60.
[4] 程先超,刘新泳,徐文方. 川芎嗪的结构改造及修饰[J].药学进展,2005,29(6):241-246.
[5] 陈学敏,许淑菊,马永雯. 2-羟甲基-3,5,6-三甲基吡嗪的合成及其对血液流变性的影响[J].中国药物化学杂志,1996,6(4):254-256.
[6] Liu X Y, Zhang R , Xu W F ,etal. Synthesis of the novel liqustrazine derivatives and their protective effect on injured vascular endothelial cell damaged by hydrogen peroxide[J].Bioorg Med Chem Lett,2003,13:2123-2126.
[7] 刘新泳,徐文方,张蕊,等. 川芎醇酯类衍生物及其制备方法和含有川芎醇酯类衍生物的药物组合物与应用[P].CN ZL02 135 989X,2005.
[8] 叶云鹏,王世英. 川芎嗪的代谢转化和结构改造研究[D].北京:中国协和医科大学&中国医学科学院,1993.
[9] 刘新泳,葛蔚颖,徐丽君,等.川芎嗪体内代谢产物的合成[J].山东医科大学学报,1997,35(1):801.
[10] 赵永海,马逢时,何勇,李家明. (E)-3-{4-[ (3,5,6-三甲基吡嗪-2-基)甲氧基]-芳基}丙烯酸的合成[J].合成化学,2008,16(4):487-489.
[11] 在三颈瓶中依次加入2-羟甲基-3,5,6-三甲基吡嗪 1.52 g(10 mmol),对羟基苯甲醛1.46 g(12 mmol)和甲苯25 mL, N2保护,搅拌下缓慢滴加DCC(二环己基碳二亚胺)2.48 g(12 mmol, 1.87 mL),于130 ℃分水反应24 h(TLC跟踪)。冷却至室温,加冰水5 mL,过滤,去除生成的二环己基脲(DCU),滤液依次用5%NaOH溶液、饱和食盐水洗涤,无水MgSO4干燥,减压蒸除溶剂后经硅胶柱层析[洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=8 ∶1]分离得淡黄色固体4-[(3,5,6-三甲基吡嗪-2-基)甲氧基]苯甲醛,收率66.3%.
