miR-210对血管内皮细胞VEGF-Notch信号通路的调控*
2011-11-20娄远蕾高法梁阮琼芳崔苏萍邓志锋
娄远蕾, 刘 芬, 高法梁, 阮琼芳, 崔苏萍, 邓志锋△, 汪 泱△
(南昌大学1第一附属医院泌尿外科研究所,2第二附属医院神经外科,江西 南昌 330006)
miR-210对血管内皮细胞VEGF-Notch信号通路的调控*
娄远蕾1▲, 刘 芬1▲, 高法梁2, 阮琼芳1, 崔苏萍1, 邓志锋2△, 汪 泱1△
(南昌大学1第一附属医院泌尿外科研究所,2第二附属医院神经外科,江西 南昌 330006)
目的: 观察过表达miR-210对血管内皮细胞中血管内皮生长因子(VEGF)-Notch信号通路相关分子表达的影响,探讨miR-210调控血管新生的分子机制。方法采用常规方法培养人脐静脉内皮细胞(HUVE-12)。通过转染LV- miR-210-GFP重组慢病毒载体上调内皮细胞miR-210表达,实验分为miR-210过表达组(LV- miR-210-GFP)和对照组(LV-GFP)。采用real-time PCR检测miR-210的表达变化,流式细胞术检测ephrin-A3的表达,采用real-time PCR、Western blotting、免疫荧光细胞化学染色法分别检测VEGF-Notch信号通路相关分子VEGF、VEGF 2(VEGFR 2)、Notch1的mRNA和蛋白表达水平。结果Real-time PCR结果显示,与对照组相比,miR-210过表达组miR-210表达水平上调了(32.10±1.26)倍;流式细胞术结果显示,miR-210过表达组阳性细胞率[(73.22±1.45)%]较对照组[(12.52±0.67)%]明显增加(Plt;0.05)。Real-time PCR结果显示,miR-210过表达组 VEGF、VEGFR2、Notch1 mRNA分别较对照组上调了(7.40±0.67)、(2.50±0.10)、(9.70±0.72)倍,Plt;0.05;免疫荧光结果显示,miR-210过表达组VEGF和VEGFR2红色荧光信号均显著强于对照组(Plt;0.05)。Western blotting 结果显示,miR-210过表达组血管内皮细胞中Notch1的蛋白表达量(4.22±0.60)较对照组明显升高(Plt;0.05)。结论miR-210过表达可显著上调VEGF-Notch信号通路分子的表达水平。
miR-210; 血管内皮细胞; 血管内皮生长因子; Notch1
缺血性损伤是器官衰竭的主要诱因,尽快促进血管新生、恢复有效血供是修复损伤的关键。血管新生受多种细胞因子的调控,主要是一些促血管生长因子,如血管内皮生长因子(vascular endothelial growth factor,VEGF)家族、成纤维细胞生长因子(fibroblast growth factor,FGF)家族、肝细胞生长因子、血管生成素、内皮素-1、转化生长因子-α等,其中VEGF是重要的调控因子[1-3]。研究表明,VEGF可诱导动脉内皮细胞上Notch1 的表达,参与新生血管的形成[4]。但VEGF-Notch信号通路的上游调控因子仍所知甚少。微小RNA(microRNA,miRNA)对血管新生亦有重要的调控作用。研究报道,miR-210是缺氧诱导miRNA,在缺氧条件下,miR-210在内皮细胞的表达上调,并能促进毛细血管样结构形成及细胞迁移[5]。生物学软件预测miR-210可调控VEGF[6],然而miR-210对血管新生可能的调控途径及机制尚不清楚。
为进一步探讨miR-210调控血管新生的机制,本研究通过过表达血管内皮细胞miR-210,观察VEGF-Notch信号通路相关分子的变化,以明确miR-210对VEGF-Notch信号通路的影响。
材 料 和 方 法
1材料
1.1细胞 人脐静脉内皮细胞(human umbilical vein endothelial cells,HUVE-12)购自长沙赢润生物技术有限公司。
1.2主要试剂 Trizol reagent(Invitrogen);逆转录试剂盒(Promega);PE标记Ⅱ抗(Abcam);VEGF抗体、血管内皮生长因子受体2(vascular endothelial growth factor receptor 2,VEGFR2)抗体、Notch1抗体、ephrin-A3抗体和罗丹明标记Ⅱ抗均购自Santa Cruz。
2方法
2.1细胞培养 以1×109/L HUVE-12细胞接种于培养瓶中,在含15%FBS的RPMI-1640培养液中置于37 ℃、5%CO2孵箱中培养,隔天换液。细胞贴壁生长呈鹅卵石样;细胞增殖至约80%融合时,0.25%EDTA胰蛋白酶消化传代。
2.2重组慢病毒的构建及转染 针对目标序列,设计合适的PCR引物;将合成的2条引物,进行退火,将目标载体与引物退火产物分别进行双酶切。将纯化回收的酶切产物进行定向连接,其产物转化细菌感受态细胞,对长出的克隆先进行PCR鉴定,其上、下游引物都设计在载体上,PCR鉴定为阳性克隆,证明目的片段已经定向连入目的载体。再对PCR鉴定阳性的克隆进行测序和分析比对,比对正确的即为构建成功的质粒载体。
转染前24 h以3.0×108/L密度将内皮细胞种至6孔板。以感染复数(multiplicity of infection,MOI)为10加入0.75 μL病毒原液(终浓度为3.0×109cell/L),每孔中加入Polybrene,终浓度为5 mg/L。转染72 h后荧光显微镜下观察GFP绿色荧光,估算细胞转染效率。实验分为miR-210过表达组(LV- miR-210-GFP转染组)与对照组(LV-GFP转染组)。
2.3实时定量PCR法检测miR-210及VEGF、VEGFR2、Notch1基因的表达变化 转染72 h后收集细胞,采用Trizol法抽提总RNA,核酸测定仪测A260/A280,要求在1.8-2.0之间,同时1%琼脂糖凝胶电泳鉴定RNA的质量。参考文献[7],采用stem-loop引物进行miR-210逆转录,反应条件如下:16 ℃ 30 min,30 ℃,30 s,42 ℃,30 s,50 ℃,1 s,共60个循环;70 ℃ 15 min。以U6为内参照。普通基因表达检测以GAPDH为内参照。采用SYBR Green I qPCR方法进行扩增,检测系统为ABI 7500 real-time PCR system,反应条件如下:95℃,10 min,95 ℃,15 s,60 ℃,30 s,40个循环。最后行融解曲线分析。每个样本设3个重复管,同时设无模板阴性对照。基因表达的相对变化以2-ΔΔCt表示。
2.4流式细胞学检测ephrin-A3表达变化 消化细胞,调整密度为2.5×105cells/50 μL的PBS细胞悬液,加入多聚甲醛固定,加入Ⅰ抗2.5 μL,加入PE标记的Ⅱ抗1 μL,避光保存待上机检测。
2.5免疫荧光染色法检测VEGF和VEGFR2的表达变化 将细胞种植在盖玻片上,约生长密度在60%取出。以1%牛血清白蛋白封闭,加Ⅰ抗(1∶100),37 ℃孵育2 h,加Ⅱ抗(1∶200),37 ℃下避光反应50 min,DAPI染色5 min,50%甘油封片,置于荧光显微镜下观察并拍照。每张盖玻片选取5个不重叠的视野于同一曝光度下拍照,采用Image J软件半定量比较两组红色荧光吸光度平均值。
2.6Western blotting检测Notch1的表达变化 收集内皮细胞,按照总蛋白提取试剂盒说明提取总蛋白并进行蛋白定量。取30 μg蛋白上样,PAGE电泳,转至硝酸纤维素膜2 h,5%脱脂奶粉室温封闭1 h。加Ⅰ抗(1∶1 000大鼠抗人Notch1),37 ℃孵育2 h,TBST缓冲液洗膜,加入辣根过氧化物酶标记的山羊抗大鼠IgG(1∶3 000),37 ℃孵育1 h,BST缓冲液洗膜,ECL试剂显影。以β-actin蛋白作为内参照。
3统计学处理
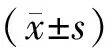
结 果
1慢病毒转染内皮细胞后miR-210表达水平明显上调
重组慢病毒转染至HUVE-12后,荧光显微镜下可见绿色荧光,GFP阳性率在80%以上。Real-time PCR 结果显示,LV- miR-210-GFP慢病毒转染内皮细胞后,与对照组比较,miR-210上调了(32.10±1.26)倍,Plt;0.05,见图1。这表明LV- miR-210-GFP的重组慢病毒转染可过表达miR-210。
2miR-210可抑制内皮细胞ephrin-A3的表达
流式细胞学结果显示,miR-210过表达组和对照组ephrin-A3阳性细胞率分别为(12.52±0.67)%和(73.22±1.45)%,miR-210转染后ephrin-A3阳性细胞明显减少,Plt;0.05,见图2。这表明miR-210的上调可抑制其靶基因ephrin-A3的蛋白表达。
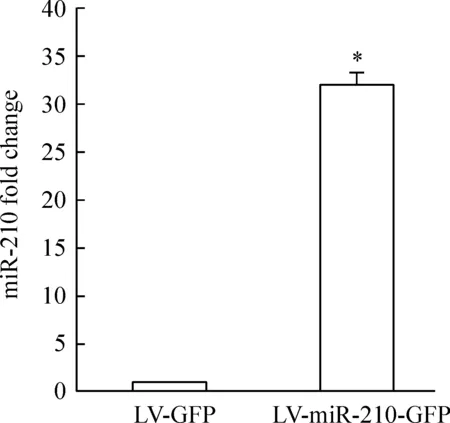
Figure 1.MiR-210 overexpression in HUVE-12 cells.miR-210 expression was significantly up-regulated after LV-miR-210-GFP transfection±s.n=3.*Plt;0.05 vs LV-GFP group.
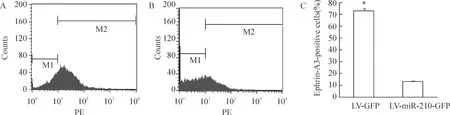
Figure 2.The expression of ephrin-A3 protein after miR-210 overexpression in vascular endothelial cells.Flow cytometry analysis showed that ephrin-A3 protein was down-regulated compared to LV-GFP group;A:LV-GFP group;B: LV- miR-210-GFP group; C:percentage of ephrin-A3-positive cells .±s.n=3.*Plt;0.05 vs LV-GFP group.
3miR-210可促进血管内皮细胞VEGF、VEGFR2、Notch1mRNA的表达
Real-time PCR 结果显示,miR-210过表达后, VEGF、VEGFR2、Notch1较对照组分别上调了(7.40±0.67)倍、(2.50±0.10)倍、(9.70±0.72)倍,Plt;0.05,见图3。表明miR-210可上调血管内皮细胞VEGF-Notch信号通路相关分子的mRNA表达水平。
4miR-210可促进血管内皮细胞VEGF、VEGFR2、Notch1蛋白的表达
免疫细胞化学荧光染色结果显示,VEGF、VEGFR2阳性红色荧光信号位于细胞胞浆和胞膜,与对照组比较,miR-210过表达组VEGF和VEGFR2红色荧光信号均显著增强,通过Image J软件半定量分析,VEGF和VEGFR2荧光吸光度值明显增加(Plt;0.05),见图4。Western blotting结果显示,与对照组比较,miR-210过表达组血管内皮细胞中Notch1的蛋白表达条带信号增强,通过Image J软件半定量分析,miR-210过表达组蛋白表达量为4.22±0.60,对照组蛋白表达量为1,两组比较有显著差异(Plt;0.05),见图5。
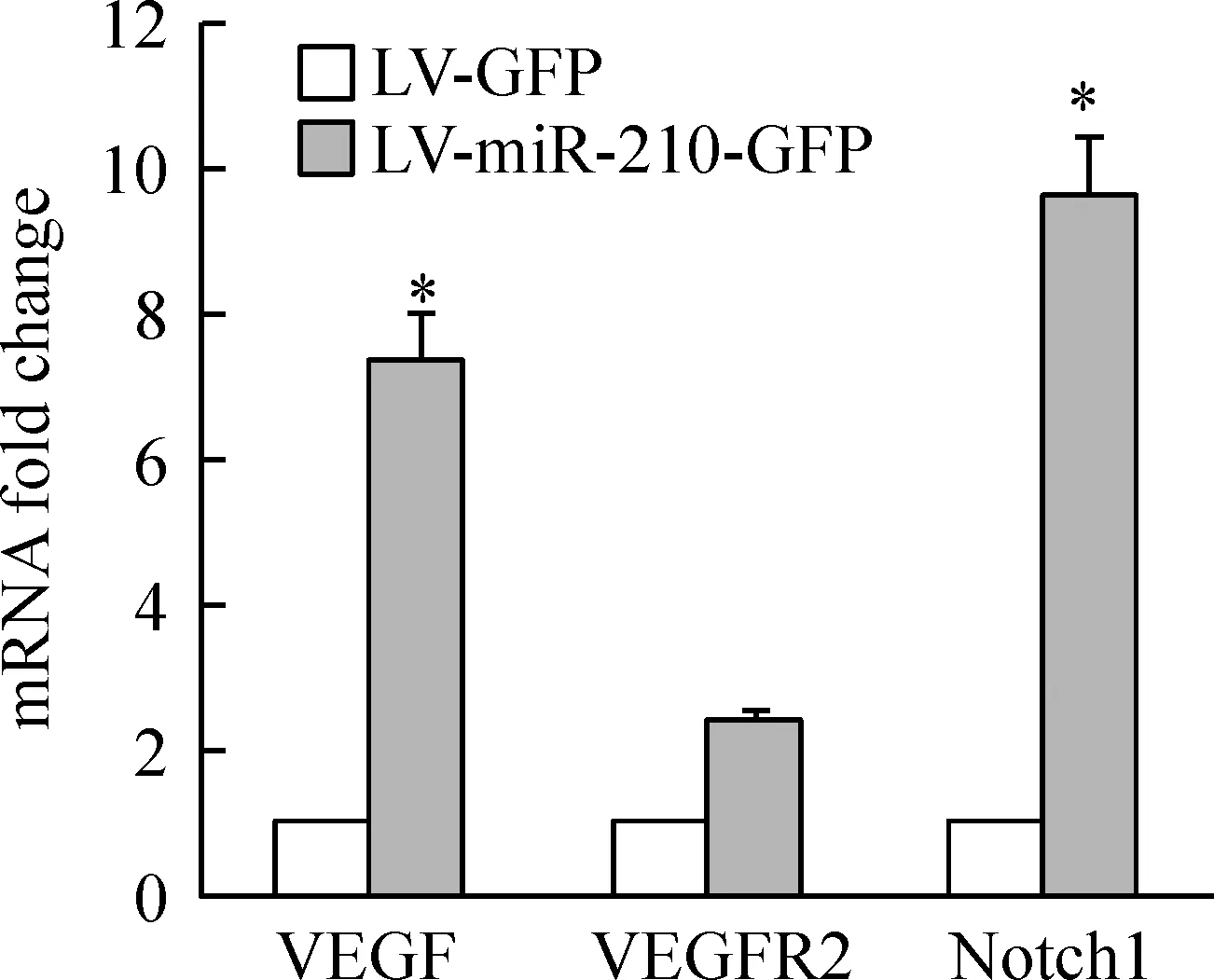
Figure 3.The expression of VEGF,VEGFR2 and Notch1 mRNA after miR-210 overexpression in vascular endothelial cells.RT-PCR showed that mRNA expression of VEGF,VEGFR2 and Notch1 was all up-regulated compared to LV-GFP group .±s.n=3.*Plt;0.05 vs LV-GFP group.
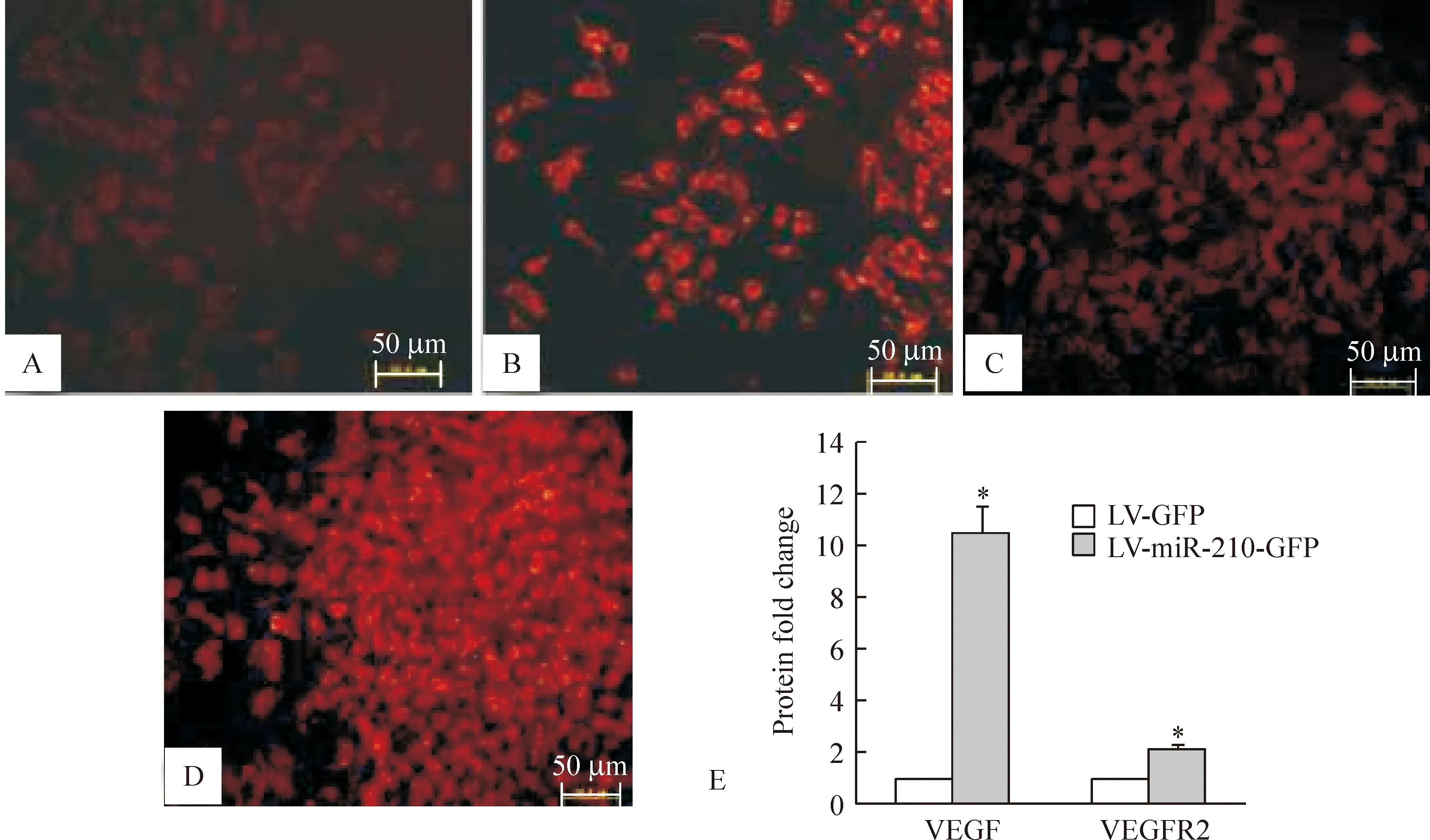
Figure 4.The protein expression of VEGF and VEGFR2 after miR-210 overexpression in vascular endothelial cells.The result of immunofluorescence showed the protein expression of VEGF and VEGFR2 was up-regulated compared to LV+GFP group.A:VEGF (LV-GFP group);B: VEGF(LV- miR-210 -GFP group);C: VEGFR2(LV-GFP group);D: VEGFR2 (LV- miR-210 -GFP group).Bar=50um.E:VEGF and VEGFR2 protein fold change±s.n=3.*Plt;0.05 vs LV+GFP group.
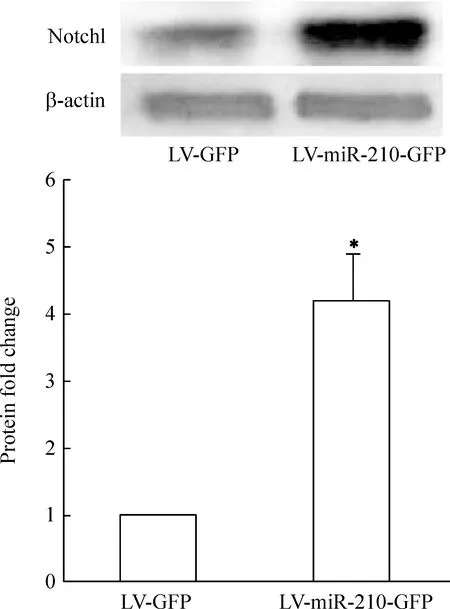
Figure 5.The expression of Notch1 after miR-210 overexpression in vascular endothelial cells.Western blotting showed that Notch1 was up-regulated compared to LV+GFP group±s.n=3.*Plt;0.05 vs LV+GFP group.
讨 论
近年研究表明,miRNA对血管新生具有重要的调控作用[8]。众所周知,脏器缺血可诱发反应性血管新生(angiogenesis),即在原有血管的基础上,内皮细胞增殖、迁移和重塑形成新血管[9]。通过促血管新生来修复缺血性损伤已成为国内外新的研究热点。研究发现,miR-210是缺氧诱导miRNA。缺氧时,miR-210在内皮细胞中的表达上调,并促进毛细血管样结构形成及细胞迁移,而抑制miR-210能阻断血管新生[5]。我们的前期研究表明,脑缺血后皮质区miR-210表达稳定上调[10];小鼠肾缺血后miR-210亦表达上调,其靶基因ephrin-A3蛋白显著下调[11],但miR-210对缺血/缺氧后血管新生可能的调控机制尚不清楚。故本研究通过在血管内皮细胞过表达miR-210,探讨其对血管新生相关信号分子的影响。
在血管生成过程中,VEGF是最重要的调控因子。研究表明,VEGF作为Notch信号的上游调控因子,可诱导动脉内皮细胞上Notch1和DLL4(Notch配体)的表达,从而促进血管的发育[4]。miR-210能否对VEGF-Notch信号通路起调控作用,对于阐明缺血后血管新生的机制具有重要意义。
通过生物学软件预测,miR-210可调控VEGF[6]。有研究证实miR-210可影响内皮细胞对VEGF的敏感性[5]。但miR-210对Notch信号分子的影响尚未见报道。本研究过表达血管内皮细胞的miR-210,观察VEGF-Notch信号通路相关分子表达的变化。研究结果显示,与对照组比较,miR-210过表达组miR-210显著上调,其靶基因ephrin-A3的阳性细胞数明显减少,证实LV- miR-210-GFP重组慢病毒载体可介导稳定过表达miR-210; VEGF、VEGFR2、Notch1基因及蛋白表达水平较对照组显著上调,说明miR-210过表达可显著上调VEGF-Notch信号通路相关分子的表达水平。提示miR-210可通过VEGF-Notch信号通路参与血管新生的调控过程,为阐明miR-210调控血管新生的分子机制提供了实验依据。
[1]Yancopoulos GD.Vascular-specific growth factors and blood vessel formation[J].Nature,2000,407(6801):242-248.
[2]张允岭,刘 超,刘雪梅.心脑舒通胶囊对局灶性脑缺血再灌注大鼠皮层VEGF、VEGFR2 mRNA表达的影响[J].中国病理生理杂志,2010,26(3):587-589,592.
[3]王 艳,邵建华.腺苷促进人脐静脉内皮细胞bFGF的表达[J].中国病理生理杂志,2005,21(5):882-885.
[4]Lawson ND,Vogel AM,Weinstein BM,et al.Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation[J].Dev Cell,2002,3(1): 127-136.
[5]Fasanaro P,D’Alessandra Y,Di Stefano V,et al.Microrna-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand ephrin-A3[J].J Biol Chem,2008,283(23):15878-15883.
[6]Hua Z,Lv Q,Ye W,et al.MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia[J].PLoS One,2006,1: e116.
[7]Varkonyi-Gasic E,Wu R,Wood M,et al.Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs[J].Plant Methods,2007,3(10):1-12.
[8]Ambros V.The functions of animal microRNAs[J].Nature,2004,431(7006):350-355.
[9]Basile DP.The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function[J].Kidney Int,2007,72(2): 151-156.
[10]高法梁,汪 泱,娄远蕾,等.缺血性脑损伤后MicroRNA 210及其靶基因ephrin-A3的表达变化[J].中华实验外科杂志,2010,27(2):264.
[11]邓 君,崔苏萍,汪 泱,等.肾缺血再灌注损伤后miR-210及其靶基因的变化[J].中华实验外科杂志,2009,26(11):1512-1515.
控制人类诱导多能干细胞多能性和决定早期细胞命运的信号通路
胚胎源性人类多能干细胞和重编程的(reprogrammed)体细胞来源的人类多能干细胞的共同特征包括无限增殖和持久、广泛地向各种类型细胞分化的潜能。但是在维持细胞多能性和调控细胞分化方面,这两种来源的细胞是否依赖相似的机制仍然存在争议,而在这些机制中出现任何一点差异都将会导致经重编程的成纤维细胞产生的多能诱导干细胞(induced pluripotent stem cells,iPSCs)的多能性发生异常改变。在此情况下,目前用于促使人类胚胎干细胞(embryonic stem cells,ESCs)扩增和分化的方法可能并不能直接适用于人类iPSCs的扩增和分化。欧洲科学家发现,人类iPSCs依赖activin/nodal/TGFβ信号通路,控制同源蛋白转录因子Nanog表达,从而维持其多能性(人类ESCs也同样依赖该信号通路)。由此可见,人类iPSCs维持自身多能性的机制与人类ESCs的机制相似。同时,他们还证实,在人类ESCs特异地分化为胚外组织、神经外胚层及中胚层的过程中,充足的生长因子是其必要条件,而这些生长因子同样可以促使人类iPSCs分化为以上各组织。以上实验为了避免可能出现的干扰发育机制分析的因素,是在纯粹的化学合成的培养基中进行的,而且实验获得的某些组织与今后临床应用需要的并不完全相同。综上所述,人类iPSCs维持自身多能性和调控自身分化所依赖的机制与人类ESCs的机制相似,也可以说这些不同类型的多能细胞在功能方面是等同的。
Stem Cells,2009,27(11):2655-2666(李丽娜)
RegulatoryeffectofmiR-210overexpressiononVEGF-Notchsignalingpathway
LOU Yuan-lei1,LIU Fen1,GAO Fa-liang2,RUAN Qiong-fang1,CUI Su-ping1,DENG Zhi-feng2,WANG Yang1
(1InstituteofUrology,TheFirstAffiliatedHospital,2DepartmentofNeurosurgery,TheSecondAffiliatedHospital,NanchangUniversity,Nanchang330006,China.E-mail:wangy63cn@sina.com)
AIM: To explore the potential mechanism of angiogenesis by investigating the regulatory effect of miR-210 overexpression on vascular endothelial growth factor(VEGF)-Notch signaling pathway.METHODSMiR-210 overexpression was induced in human umbilical vein endothelial(HUVE-12) cells by lentivirus transfection.The HUVE-12 cells were divided into 2 groups: miR-210 overexpression group (LV-miR-210-GFP) and control group (LV-GFP).The change of ephrin-A3 protein was detected by flow cytometry analysis.The mRNA and protein levels of VEGF-Notch signaling molecules,VEGF,VEGF receptor 2(VEGFR 2)and Notch 1,were determined by the methods of real-time PCR,immunofluorescence and Western blotting.RESULTSCompared to the control cells,significant down-regulation of ephrin-A3 protein was observed in HUVE-12 cells with miR-210 overexpression,while both mRNA and protein levels of VEGF,VEGFR2 and Notch1 were up-regulated.CONCLUSIONmiR-210 may be involved in VEGF-Notch signaling pathway to regulate angiogenesis.
miR-210; Vascular endothelial cells; Vascular endothelial growth factors; Notch1
1000-4718(2011)05-0923-05
R363
A
10.3969/j.issn.1000-4718.2011.05.017
2010-11-24
2011-03-09
国家自然科学基金资助项目(No.30960396;No.81060059)
△通讯作者 邓志锋 Tel:0791-6282662;E-mail:dengzf@126.com; 汪泱 Tel:0791-8692527;E-mail:wangy63cn@sina.com
▲并列第1作者
