基于对羟基苯乙酸及邻菲咯啉的稀土配合物的合成、晶体结构和荧光光谱
2011-11-09刘建风陈吉妃赵国良
刘建风 陈吉妃 赵国良*,,2
(1浙江师范大学化学与生命科学学院,金华 321004)(2浙江师范大学行知学院,金华 321004)
基于对羟基苯乙酸及邻菲咯啉的稀土配合物的合成、晶体结构和荧光光谱
刘建风1陈吉妃1赵国良*,1,2
(1浙江师范大学化学与生命科学学院,金华 321004)(2浙江师范大学行知学院,金华 321004)
在乙醇-水体系中合成了5个对羟基苯乙酸及邻菲咯啉稀土配合物:其中4个配合物[RE(HPAA)3(phen)2]·2H2O(RE=Eu(1),Tb(2),Dy(3),Yb(4))具有相同的结构和类似的化学组成;第 5 个配合物是[Yb(HPAA)2(H2O)2(phen)2](HPAA)·(HHPAA)·2H2O(5),HHPAA=对羟基苯乙酸,C8H8O3;phen=1,10-邻菲咯啉),其结构和化学组成与前4个配合物不同。并通过元素分析、红外光谱、热重分析和粉末X-射线衍射对产物进行表征,用单晶X-射线衍射方法测定了配合物5的晶体结构。配合物5(C56H53N4O16Yb)属于三斜晶系,空间群 P21/c,晶胞参数:a=2.20652(3)nm,b=1.36876(2)nm,c=1.75414(2)nm,β=101.167(1)°,晶胞体积:V=5.19754(12)nm3,晶胞内结构基元数Z=4,分子量Mr=1211.06。测定了铕、铽和镝配合物的荧光光谱,结果表明,在形成配合物后,依然显示铕(Ⅲ)离子、铽(Ⅲ)离子和镝(Ⅲ)离子的特征发射,这表明配体将吸收的能量有效地转移给了中心离子,配体起到了很好的敏化作用。
稀土配合物;对羟基苯乙酸;邻菲咯啉;晶体结构;荧光光谱
稀土化合物具有光、电、磁三大功能,其中最突出的功能是发光,在稀土应用研究当中,发光材料占主要地位[1]。稀土化合物之所以具有发光的功能是因为它的原子或离子具有丰富的能级和4f电子,其光谱中可检测的谱线多达30000条,其中涵盖了从紫外光区到红外光区的多种电子能级,为多种不同能量的能级跃迁创造了条件,有着其他元素和化合物无法比拟的优势。特别是铕、铽、镝、钐等元素发光能级位于可见光范围并且发光频率不同,使得稀土材料成为研究新型发光材料和发光机理的理想原料[2]。从世界范围看,稀土化合物作为发光材料的研究和应用已经有100多年的历史了,目前依然方兴未艾,稀土发光材料在农业[3-4]、生物技术[5]、电子元件[6]及采矿等多个领域中都有着广泛的应用前景。近年来对稀土配合物发光材料的研究发展迅速,由于稀土配合物兼有稀土离子的多样能级跃迁现象和有机物的紫外光区的宽频吸收特性,所以有机配合物可以结合稀土与配体各自的优点,扬长避短以得到光效更高更加方便易得的新型发光材料。研究表明稀土配合物所发出的荧光既有稀土离子发光强度高、颜色纯正的优点,又兼具有机化合物的激发能量低、荧光效率高、易溶于有机溶剂的特点。一般选择可以吸收紫外光的有机物作为配体,结合在稀土离子上能起到天线效应,选择跃迁能级适合的配体可以有效地将能量传递给中心离子,达到增强发光效率的目的,这就为人们探索新的发光能源和发光材料提供了新的思路。正因为配合物与传统稀土掺杂氧化物荧光粉相比具有更高的发光效率和更窄的谱带宽度,因此其应用价值和发展前景更受瞩目[7],此后大量有关稀土配合物荧光现象、机理及应用的研究在不同领域展开[8-20]。本文合成了对羟基苯乙酸邻菲咯啉与铕、铽、镝、镱(Ⅲ)的配合物[RE(HPAA)3(phen)2]·2H2O(RE=Eu(1),Tb(2),Dy(3),Yb(4),[Yb(HPAA)2(H2O)2(phen)2](HPAA)·(HHPAA)·2H2O(5),HHPAA=对羟基苯乙酸,C8H8O3;phen=1,10-邻菲咯啉),并通过元素分析、红外光谱、热重分析和粉末X-射线衍射对产物进行表征,用单晶X-射线衍射方法测定了配合物5的晶体结构,并测定了铕、铽和镝配合物的荧光光谱。
1 实验部分
1.1 试剂和仪器
对羟基苯乙酸、邻菲咯啉、RE(NO3)3·6H2O均购自济南恒化科技有限公司,分析纯。无水乙醇等试剂均为市售分析纯试剂,用前未做进一步处理。
德国Elementar公司Vario ELⅢ型元素分析仪;美国Nicolet公司NEXUS 670型傅立叶变换红外光谱仪(FTIR),KBr压片,测定范围为 400~4 000 cm-1;上海雷磁仪器厂的DDS-11A型电导率仪;英国Edinburgh公司FL 920荧光光谱仪;瑞士METTLER-TOLEDO公司TGA/SDTA 851e型热分析仪;德国Bruker公司APEXⅡCCD单晶衍射仪;荷兰Philips公司PW3040/60X′pert Pro全自动Cu Kα X-射线衍射仪。
1.2 配合物的合成
分别将 Eu(NO3)3·6H2O、Tb(NO3)3·6H2O、Dy(NO3)3·6H2O和Yb(NO3)3·6H2O各1 mmol溶于5 mL蒸馏水中,再加入10 mL浓度为1 mol·dm-3的对羟基苯乙酸钠水溶液并不断搅拌,用1 mol·dm-3稀硝酸调节溶液为弱酸性 (pH值约为5),15 min后滴加1 mmol邻菲咯啉的乙醇溶液(10 mL),有白色沉淀产生,继续搅拌1 h。将沉淀过滤,用乙醇洗涤后放入干燥器中干燥。让所有滤液在室温下自然挥发溶剂,30 d以后,配合物4的滤液中长出无色柱状晶体得配合物5,其他各滤液未得到单晶。配合物1、2、3、4 粉末产率在 21%~54%之间 (以 RE(NO3)3·6H2O计)。对4个粉末样和1个单晶样进行元素分析,C48H41Eu N4O11(1)(Mr=1 001.81),实验值(%):C,57.14;H,4.16;N,5.56。计算值(%):C,57.54;H,4.13;N,5.59。C48H41TbN4O11(2)(Mr=1 008.77),实验值(%):C,56.94;H,4.12;N,5.53。计算值 (%):C,57.15;H,4.10;N,5.56。C48H41DyN4O11(3)(Mr=1012.35),实验值(%):C,56.73;H,4.13;N,5.49。计算值(%):C,56.94;H,4.08;N,5.54。C48H41YbN4O11(4)(Mr=1022.89),实验值(%):C,56.43;H,4.09;N,5.39。计算值(%):C,56.36;H,4.04;N,5.48。C56H53YbN4O16(5)(Mr=1211.06),实验值(%):C,55.31;H,4.37;N,4.71。 计 算 值 (%):C,55.53;H,4.41;N,4.63。
1.3 晶体结构测定及解析
选用大小为 0.071 mm×0.069 mm×0.046 mm 的配合物5单晶,在德国Bruker SMART APEXⅡCCD单晶衍射仪上上进行衍射实验。用辐射Mo Kα射线(λ=0.071 073 nm),在设定的 2θ角范围内收集衍射数据。衍射数据经Lp因子和经验吸收校正[21]。晶体结构由直接法[22]解出。所有非氢原子的坐标用全矩阵最小二乘法进行各向温度因子修正。在各向异性化温度因子修正后,对碳原子进行理论加氢,水上的氢通过Fourier合成得到,并对键长作限制性精修(d(O-H)=0.085 nm,d(H-H)=0.130 nm),单晶结构解析工作在Pentium PC计算机上用SHELX-97[22]程序完成。配合物最后的残差因子R1=0.0386,wR2=0.0633。主要晶体学数据列于表1,主要的键长和键角列于表2。
CCDC:773376。
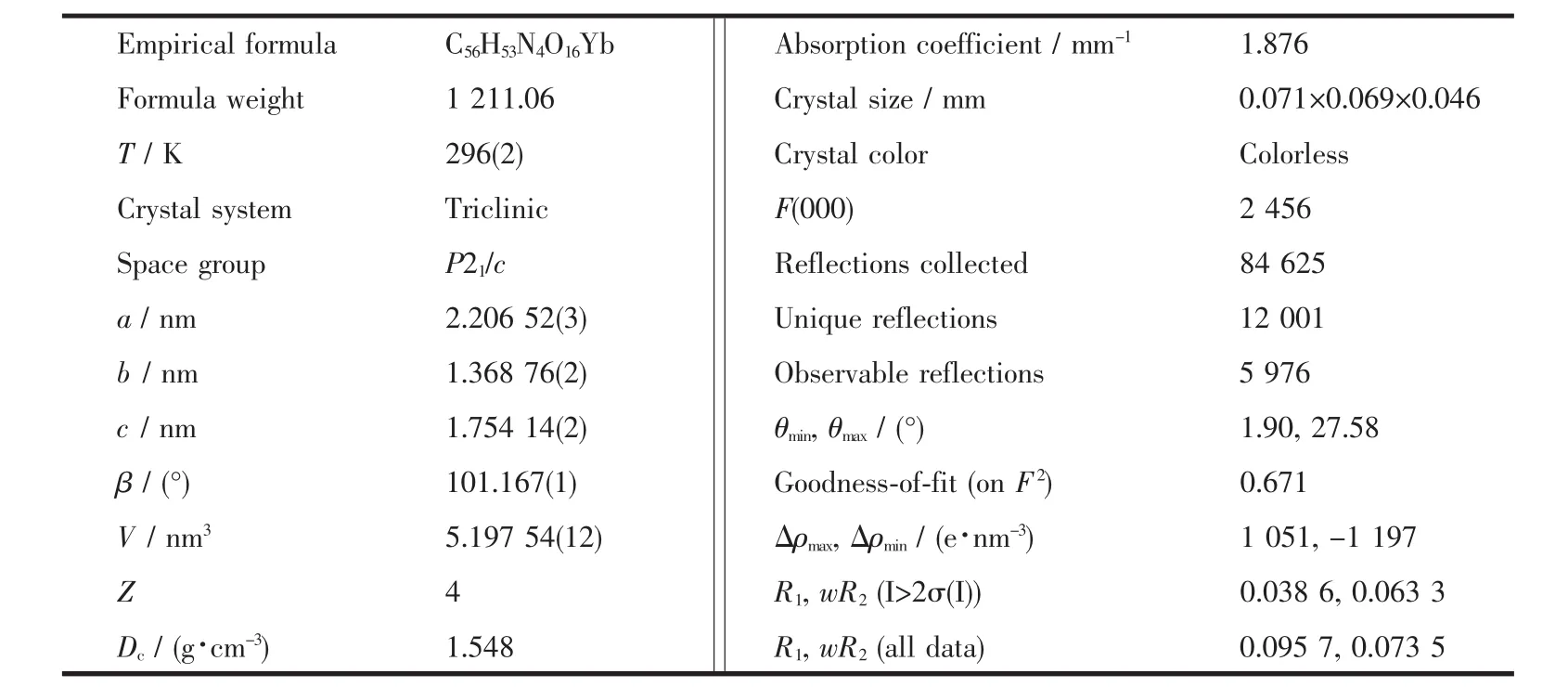
表1 配合物5的晶体学数据Table 1 Crystal data of the complex 5

表2 配合物5的主要键长和键角Table 2 Selected bond distances(nm)and bond angles(°)of the complex 5
2 结果与讨论
2.1 配合物的组成
由配合物的元素分析数据可知,配合物1、2、3、4的化学式为C48H41REN4O11,即有1个中心离子、2个邻菲咯啉分子、3个对羟基苯乙酸根离子和2个水分子所组成。25 ℃时,1.0 mmol·dm-3DMF 溶液的摩尔电导值分别为 1.8、1.9、1.2 和 1.3 S·cm2·mol-1,属于非电解质[23],是分子型配合物,再参考配合物5的晶体结构,推测它们的分子式可能为[RE(HPAA)3(phen)2]·2H2O。
2.2 配合物的XRD
图 1 为配合物 1、2、3、4 的粉末 X-射线衍射(XRD)图。由图可知,4个配合物的图很相似,说明它们具有相似的结构,从而进一步说明它们具有相似的化学组成C48H41REN4O11。
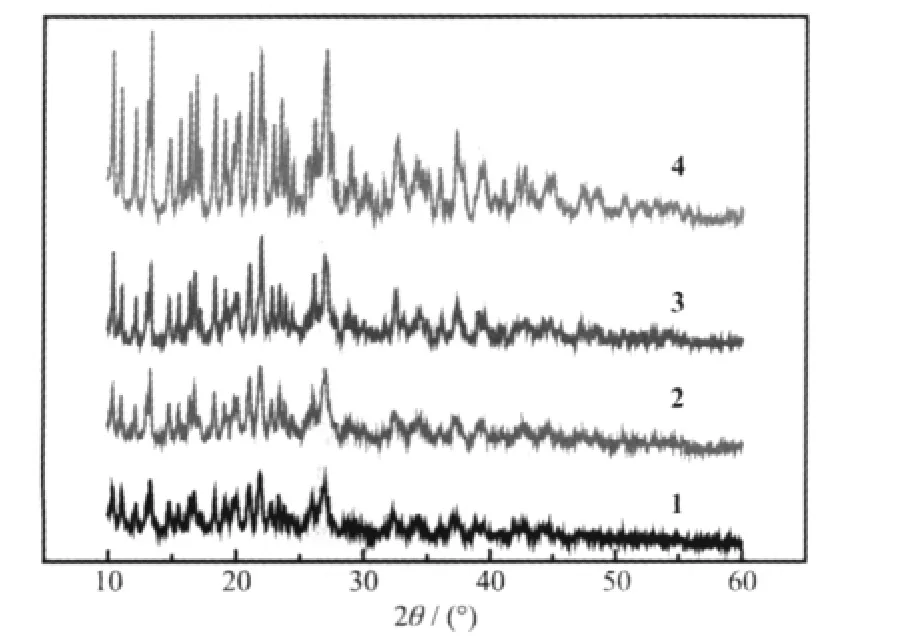
图1 配合物1、2、3、4的XRD图Fig.1 XRD of complexes 1,2,3,4
2.3 配合物的红外光谱
配合物1、2、3、4的红外光谱很相似,以配合物1为例来说明,3415 cm-1处存在宽峰,为O-H伸缩振动峰;自由配体HHPAA于1697 cm-1处的特征吸收峰νC=O(-COOH)在形成配合物后消失,而出现了羧酸根(-COO-)的反对称νas(COO-)1602 cm-1和对称伸缩振动吸收峰νa(COO-)1397 cm-1,表明羧基中的氧原子参与了配位[24-25]。邻菲咯啉在形成三元配合物后,C=N特征伸缩振动峰从1 561 cm-1红移至1 516 cm-1,面外变角振动δC-H从739 cm-1移至为729 cm-1,δC-C也从 853 cm-1向低波数方向 848 cm-1移动,表明邻菲咯啉中的2个氮原子同时参与配位[26]。对于配合物5来说,由于其化学组成与1、2、3、4有所不同,反映在红外光谱上这些相应的特征伸缩振动吸收峰略有位移,邻菲咯啉的特征吸收峰分别出现在1516、843、728 cm-1处;配位羧酸根的反对称νas(COO-)和对称νa(COO-)伸缩振动吸收峰分别出现在1 594和1 394 cm-1,归属于自由配体HHPAA的于 1 697 cm-1处的特征吸收峰 νC=O(-COOH)依旧存在,表明配合物中有未电离的游离配体HHPAA存在,这与晶体结构的测定结果一致。
2.4 配合物的晶体结构
图2为配合物5的分子结构图,从图中可以看出,配合物是零维的配合物分子,其晶体结构的最小不对称单元中包括1个中心金属离子、2个phen分子、4个水分子、3个对羟基苯乙酸根HPAA-离子和1个游离的对羟基苯乙酸HHPAA分子。配合物的内界为+1价配位阳离子,其中心金属离子Yb(Ⅲ)为八配位,呈略有畸变的反四方棱柱体构型,如图3所示。2个phen分子与Yb3+双齿螯合,4个Yb-N键长为 0.246 3(3)~0.252 7(4)nm,与已报道的八配位Yb配合物类似[26]。2个phen分子平面所形成的夹角为 35.020(0)°。2 个 HPAA-分别以其羧基上的 1 个氧与Yb3+单齿配位,Yb与O之间配位键长分别为Yb-O1 0.227 5(3)nm,Yb-O4 0.229 1(3)nm,这与文献[27]报道的八配位Yb配合物基本相同。HPAA-上2个苯环所确定的最小二平面之间的夹角为46.557°。剩余2个配位点被2个配位水占据,水与Yb 之间的键长为 Yb-O(1w)=0.2297(3)nm,Yb-O(2w)=0.2243(3)nm,与结构类似的Yb配合物接近[28]。在配合物的外界有1个游离的HPAA-离子使整个配合物晶体仍呈电中性。另外晶格中还存在1个未电离的HHPAA分子和2个水分子,通过H键将整个分子联系在一起,配合物的氢键数据列于表3。
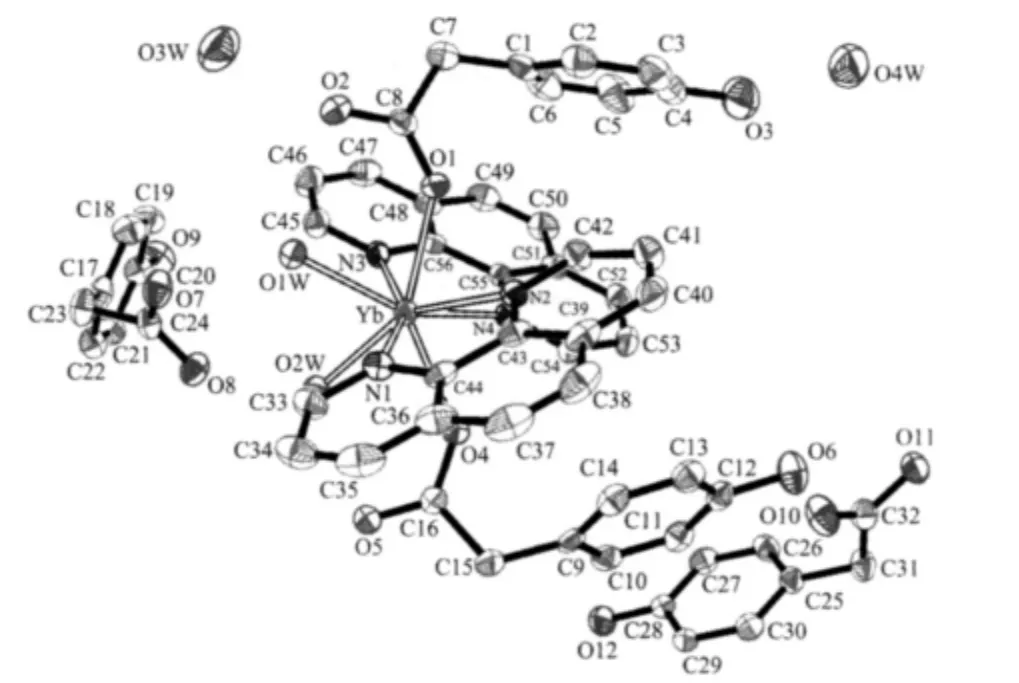
图2 配合物5的分子结构图Fig.2 Molecular structure of the complex 5,ellipsoids are shown at the 30%level

图3 配合物5中Yb的配位环境图Fig.3 Coordination environment of the Yb of the complex 5
图4是配合物5沿c轴方向排列的分子堆积图,从中可以明显地看到晶体中存在一些方形的孔道和狭长的缝隙,这些方形孔道被游离的对羟基苯乙酸根HPAA-离子所填充,孔道中的HPAA-离子通过H键首尾相接,每个羧基氧都作为受体与周围的活泼H形成4个氢键,在孔道中起着固定和导向的作用。狭长的层状缝隙宽度为0.150 nm,与1个氧原子的范德华半径相当,缝隙的两侧是参与配位的HPAA-离子,同侧相邻2个HPAA-的苯环平面几乎平行,二面角为10.534°,游离水分子就填充在HPAA-围成的缝隙之间,每个水分子都借助自己的2个氢作为供体和1个氧作为受体形成3个氢键,并在ab面上形成了椅式的六元氢键环,2个六元氢键环被1个菱形的四元氢键环连接起来。这些氢键将配合物连接成沿bc面无限扩展的双层的配合物超分子体系,中间即为缝隙中的水层。层状体系边界为对羟基苯乙酸HHPAA分子群,它们也呈层状排列,苯环间只存在弱的疏水相互作用力和范德华力。相邻邻菲咯啉间存在π-π作用力,芳香环间质心距最短距离为0.35687(0)nm。以上这些氢键和π-π堆积等弱相互作用力对晶体的形成和稳定起着至关重要的作用。

表3 配合物5氢键数据表Table 3 Hydrogen bond data of the complex 5
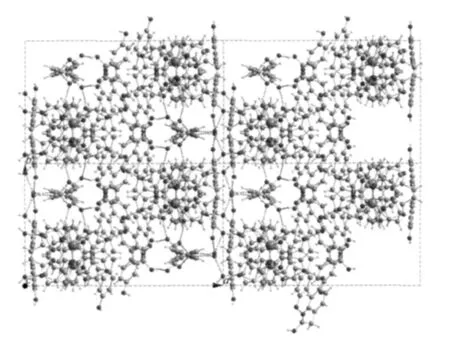
图4 配合物5的分子堆积图Fig.4 Packing plot of the complex 5
2.5 配合物的热重分析
测试温度为30~800℃,空气气氛,升温速率为10 ℃·min-1。
配合物1、2、3、4的热分解情况非常相似,现以配合物4为例来说明,图5为配合物4的TG-DTG曲线,从图中可知,配合物在第一阶段即在97℃以前失重率为3.42%,可推测为失去2个结晶水(理论值为 3.52%);第二阶段 (200~288℃)失重率为9.27%,可推测为失去 1个苯酚基 (理论值为9.10%),第三阶段(288~490 ℃)失重率为 67.14%,该阶段样品灰化,对应失去其余所有的配体(但保留氧化镱所需的氧,理论失重值68.02%),残余量20.17%,与 Yb2O3基本相当(理论残余 19.26%)。图 6是配合物5的TG-DTG曲线图,从图可知配合物的热分解过程主要有3个阶段:第一阶段,132℃前共失去总质量的6.17%,对应配合物分子中失去4个水分子(理论失重率 5.95%);第二阶段,154~244 ℃范围内的实验失重率为7.33%,与失去1个苯酚基相当(理论失重率 7.77%);第三阶段,在 244~513℃之间失去总质量的69.73%,该阶段样品灰化,对应失去其余所有的配体(但保留氧化镱所需的氧,理论失重值 69.94%),残余量 16.77%,与 Yb2O3相当(理论残余16.28%)。比较图5和图6可知配合物4与配合物5的含水量不同,配合物5的脱水温度也要高于配合物4的脱水温度,这与上述的其他表征也是一致的。

图5 配合物4的TG-DTG曲线Fig.5 TG-DTG curves of complex 4

图6 配合物5的TG-DTG曲线Fig.6 TG-DTG curve of complex 5
2.6 配合物的荧光光谱

图7 对羟基苯乙酸和邻菲咯啉的激发光谱(λem=358 nm)和发射光谱(λex=309 nm)Fig.7 Excitation(λem=358 nm)and emission spectra(λex=309 nm)curves of p-hydroxyphenylacetic acid and 1,10-phenanthroline
在相同条件下测定了HHPAA和phen的激发光谱和发射光谱(见图7),HHPAA和phen的激发峰位置重合,它们的发射峰位置也大致能重合。phen的三重态能级为22123 cm-1,稀土离子Eu3+、Tb3+的最低激发态能量分别为17277,20500 cm-1,说明配体能将自身吸收的能量传递给中心稀土离子,实现中心稀土离子的特征发射。
2.6.1 配合物1的荧光光谱
配合物1的荧光激发光谱与发射光谱如图8所示。固定配合物的发射波长为618 nm在200~500 nm范围内测得激发光谱(见小图),其激发光谱上出现了配体的宽激发峰(327 nm)和三价铕离子的特征激发峰(395 nm)。在327 nm紫外光激发下,测得配合物的发射光谱,图中可以看到Eu3+的5个特征发射峰,分别对应着5D0→7Fi(i=0,1,2,3,4)的跃迁。579 nm处小尖峰对应5D0→7F0的跃迁,辐射能量最高,荧光波长最短。590、595、689、698 nm 的 4个次强峰为5D0到7F1和7F4的跃迁并且均发生了分裂。5D0到7F3的跃迁发射位于649 nm处,峰强很弱。最强峰位于618 nm处,对应5D0→7F2的跃迁,半高宽为 5.3 nm,红橙比为 4.1,红色非常纯正。

图8 配合物1的激发光谱和发射光谱Fig.8 Excitation and emission curves of the complex 1
2.6.2 配合物2的荧光光谱
配合物2的荧光激发光谱与发射光谱如图9所示。固定配合物的发射波长为544 nm在200~500 nm范围内测得激发光谱(见小图),其最佳激发峰波长为337nm。在337 nm紫外光激发下,测得配合物的发射光谱,图中可以看到Tb3+的4个特征发射峰,分别对应着5D4→7Fi(i=3,4,5,6)的跃迁。最强发射峰位于544 nm处,对应5D4→7F5的跃迁,半高宽7.1 nm。其他3个发射峰位于489、586和621 nm处,分别对应5D4到7F6,7F4和7F3的跃迁。544、489 nm处的荧光强度比值明显较小,这可能是因为5D4→7F6跃迁为电偶极跃迁,受配体场的影响很大,而5D4→7F5跃迁属于磁偶极跃迁,受配体场的影响很小的缘故。
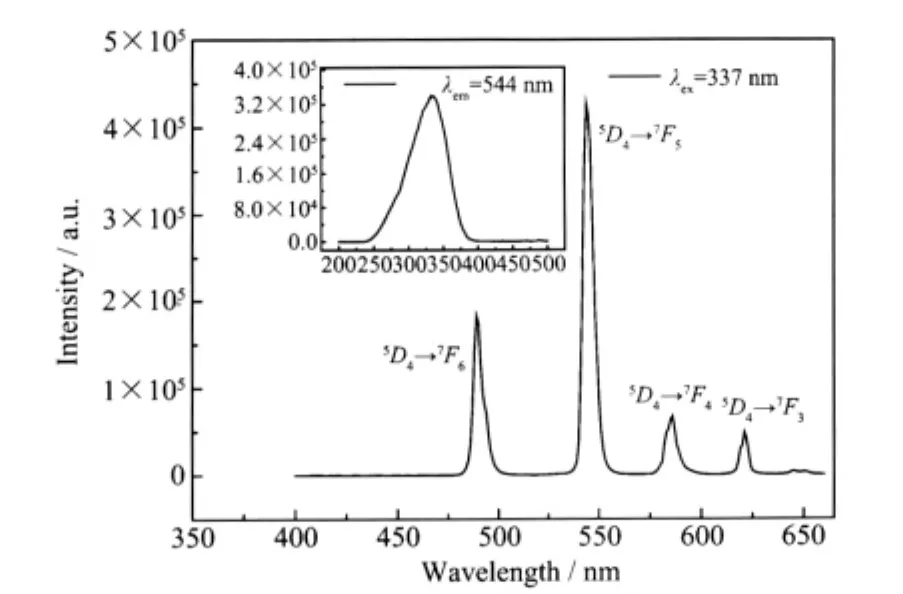
图9 配合物2的激发光谱和发射光谱Fig.9 Excitation and emission curves of the complex 2
2.6.3 配合物3的荧光光谱
配合物3的荧光激发光谱与发射光谱如图10所示。固定配合物的发射波长为575 nm在100~395 nm范围内测得激发光谱(见小图),其最佳激发峰波长为336 nm。在336 nm紫外光激发下,测得配合物的发射光谱,图中可以看到Dy3+的2个强发射峰,574(黄色)和 484 nm(蓝色),均为 Dy3+的特征发射峰,分别对应于4F9/2→6Hi(i=15/2,13/2)的跃迁。574 nm附近发射峰较强并且劈裂为2个峰,它们分别位于572和576 nm处,半高宽为10.0 nm。

图10 配合物3的激发光谱和发射光谱Fig.10 Excitation and emission curves of the complex 3
[1]XU Guang-Xian(徐光宪).Rare Earth.2nd Ed(稀土.2 版).Beijing:Metallurgical Industry Press,1978.
[2]LI Yu-Lin(李玉林).Rare Earth(Xitu),1999,20(2):67-68,72
[3]WANG Ze-Min(王则民),FU Chu-Jin(傅楚瑾),CAO Jin-Rong(曹锦荣),et al.China Plastics(Zhongguo Suliao),1999,13(12):21-23
[4]Wang J C,Liu X S,Yang J,et al.J.Rare Earths,2006,24(spec.issue):427-431
[5]Zhou M F,He Q Z.J.Rare Earths,2008,26(4):473-477
[6]Huang C H,McCaughan L.Electron.Lett.,1997,33(19):1639-1640
[7]LI Xin-Bei(李新贝),ZHANG Fang-Hui(张方辉).Advanced Displays(Xiandai Xianshi),2006,65:53-59
[8]FAN Nai-Ying(范乃英),GAO Shan(高山),HUO Li-Hua(霍丽华).Chinese J.Anal.Chem.(Fenxi Huaxue),2005,33(6):751-754
[9]LI Ling(李玲),LIU Yu-Kan(刘玉侃),TAO Jin-Hui(陶锦晖).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2008,24(9):1369-1373
[10]De Silva A P,Gunaratne H Q N,Gunnlaugsson T,et al.Chem.Rev.,1997,97(5):1515-1566
[11]MitschkeU,BuerleP.J.Mater.Chem.,2000,10(7):1471-1507
[12]WolffNE,Pressley R.J.Appl.Phys.Lett.,1963,2(8):152-154
[13]Rohatgl K K,Sen Gupta S K.J.Inorg.Nucl.Chem.,1972,34:3061-3071
[14]Horrocks J W D,Albin M.Inorg.Chem.,1984,31:1-104
[15]HUANG Chun-Hui(黄春辉).Rare Earth Coordination Chemistry(稀土配位化学).Beijing:Science Press,1997.
[16]Yang C,Chen X M,Zhang W H,et al.Dalton Trans.,1996:1767-1768
[17]QIN Li(秦利),YE Jian-Ping(叶建平),YU Qun(虞群),et al.J.Chin.Rare Earth Soc.(Zhongguo Xitu Xuebao),1991,1:28-32
[18]JIN Lin-Pei(金林培),Bünzli J C G,Plancherel D.Acta Chimica Sinica(Huaxue Xuebao),1990,48(1):58-63
[19]JIN Lin-Pei(金林培),WANG Ming-Zhao(王明昭).Science inChina:SeriesB(ZhongguoKexueBJi),1994,24(6):577-583
[20]ZHANG Hong-Jie(张洪杰),FU Lian-She(符连社),LIN Jun(林君),et al.Chin.J.Lumin.(Faguang Xuebao),2002,23(3):228-232
[21]Sheldrick G M.SADABS,Program for Empirical Absorption Correction of Area Detector Data,University of Göttingen,Germany,1997.
[22]Sheldrick G M.SHELXL 97,Program for Crystal Structure Refinemenf,University of Göttingen,Germany,1997.
[23]Geary W J.Coord.Chem.Rev.,1971,7:81-122
[24]Nakamoto K,Translated by HUANG De-Ru(黄德如),WANG Ren-Qing(汪仁庆).Infrared and Raman Spectra of Inorganic and Coordination Compounds(无机和配位化合物的红外和拉曼光谱).Beijing:Chemical Industrial Press,1986:235
[25]Liu C B,Wen H L,Tan S S.J.Mol.Struct.,2006,794:190-195
[26]Sun C H,Jin L P.J.Mol.Struct.,2005,741:241-247
[27]Liu C B,Sun C Y,Jin L P,et al.New J.Chem.,2004,28:1019-1026
[28]Baca S G,Adams H,Sykes D,et al.Dalton Trans.,2007:2419-2430
Synthesis,Crystal Structure and Fluorescence Spectrum of Rare Earth(Ⅲ)Complexes with p-Hydroxyphenylacetic Acid and 1,10-Phenanthroline
LIU Jian-Feng1CHEN Ji-Fei1ZHAO Guo-Liang*,1,2
(1College of Chemistry and Life Sciences,Zhejiang Normal University,Jinhua,Zhejiang 321004,China)(2Zhejiang Normal University Xingzhi College,Jinhua,Zhejiang 321004,China)
Fiverareearthcomplexes[RE(HPAA)3(phen)2]·2H2O(RE=Eu(1),Tb(2),Dy(3),Yb(4),[Yb(HPAA)2(H2O)2(phen)2](HPAA)·(HHPAA)·2H2O (5),HHPAA=p-hydroxyphenylacetic acid,C8H8O3;phen=1,10-phenanthroline),weresynthesized and characterized byelementalanalysis,infrared spectra,thermogravimetry-derivative thermogravimetry (TG-DTG)and X-ray powder diffraction.The crystal structure of complex 5 was determined by single crystal X-ray diffraction method.The complex,C56H53YbN4O16,crystallizes in the triclinic system,space group P21/c,with cell parameters:a=2.206 52(3)nm,b=1.368 76(2)nm,c=1.754 14(2)nm,β=101.167(1)°,cell volume:V=5.19754(12)nm3,number of molecules inside the cell:Z=4,relative molecular mass:Mr=1210.06.The excitation and emission spectra of the complexes 1,2,3 were investigated.The luminescence behaviors probably results from the ligand-sensitized metal-centered emission.CCDC:773376.
rare earth complex;p-hydroxyphenylacetic acid;1,10-phenanthroline;crystal structure;fluorescence spectra
O614.33
A
1001-4861(2011)01-0100-07
2010-05-17。收修改稿日期:2010-08-18。*
。E-mail:sky53@zjnu.cn
