配合物[Mn(EDTB)](NO3)2·DMF的合成、晶体结构及多酚氧化酶(PPO)活性的研究
2011-09-29魏丽梅裴亚梅
王 莉 魏丽梅 张 勇,2 裴亚梅
(1华中师范大学化学学院,武汉 430079)(2黄石理工学院化学与材料工程学院,黄石 435003)
配合物[Mn(EDTB)](NO3)2·DMF的合成、晶体结构及多酚氧化酶(PPO)活性的研究
王 莉*,1魏丽梅1张 勇1,2裴亚梅1
(1华中师范大学化学学院,武汉 430079)(2黄石理工学院化学与材料工程学院,黄石 435003)
合成六齿配体 N,N,N′,N′-四(2-苯并咪唑亚甲基)-1,2-乙二胺(EDTB)及单核锰(Ⅱ)配合物[Mn(EDTB)](NO3)2·DMF,并进行了晶体结构和多酚氧化酶(PPO)模拟活性研究。该配合物为正交晶系,P212121空间群,a=1.172 52(6)nm,b=1.320 83(6)nm,c=2.46191(12)nm,V=3.8128(3)nm3,Dc=1.451 g·cm-3,Z=4,F(000)=1732。最终因子 R(I>2σ(I)):R1=0.0522,wR2=0.1022;R(全部数据):R1=0.0652,wR2=0.1075,Flack 指数为 0.02(15)。结构分析表明,锰(Ⅱ)分别与配体 EDTB 苯并咪唑环的 4 个氮原子、两个亚胺基氮原子发生配位,形成1个变形八面体结构。采用分光光度法研究配合物的多酚氧化酶活性,以邻苯三酚为底物,表明在0.1~10 μmol·L-1范围内,随着配合物浓度逐渐增大,配合物的氧化促进率从 51.5%增至 82.2%。在 30 ℃,pH=7.8~8.2 条件下,配合物的转化数由22.93 h-1增加到51.01 h-1,说明配合物的PPO活性随着pH值的升高而明显增强。
锰(Ⅱ)配合物;EDTB;晶体结构;PPO活性。
0 引 言
多酚氧化酶(PPO)是一种广泛分布在微生物,植物和动物体内的氧化还原酶[1]。生物体内PPO酶儿茶酚的活性部位含双核铜,两个铜离子被羟基桥连,间距为0.287 nm,每个铜离子另外与3个组氨酸咪唑氮配位,形成四配位的四面体结构[2]。多酚氧化酶与植物的抗病性有关,植物在抵御病原微生物的侵染过程中,病原相关蛋白家族PPO和酚类代谢系统中的一些酶通过催化木质素及醌类化合物的形成,构成保护性屏蔽而使细胞免受病菌的侵害,也可以通过形成醌类物质直接发挥抗病作用[3]。此外,在工业和农业生产中,PPO的作用直接影响食品、饮料的色泽及品质,特别是在制作绿茶、红茶、烤烟和水果类饮料的过程中更为突出[4-5]。多酚氧化酶一直是研究的热点,其生化、生理学性质方面已取得了较大的进展。
本文选择六齿配体 N,N,N′,N′-四(2-苯并咪唑亚甲基)-1,2-乙二胺(C34H32N10,EDTB)为配体,合成锰-EDTB配合物作为PPO的模型配合物。采用X-射线衍射法测定了其晶体结构,并进行了元素分析、红外光谱、紫外光谱表征。采用分光光度法研究配合物的多酚氧化酶活性,以邻苯三酚为底物,表明在 0.1~10 μmol·L-1范围内,随着配合物浓度逐渐增大,配合物的氧化促进率从51.5增至82.2%.在30 ℃,pH=7.8~8.2 条件下,配合物的转化数由 22.93 h-1增加到51.01 h-1,说明配合物的PPO活性随着pH值的升高而明显增强。
1 实验部分
1.1 试剂与仪器
试剂均为市售分析纯。Perkin-Elmer 2400型元素分析仪;JEAN-Specord 210型紫外可见分光光度计;NEXUS智能型高分辨率傅里叶红外光谱仪;WS70-1红外线快速干燥器;Brucker AXS SMART 1000 CCD单晶衍射仪。
1.2 EDTB配体的合成
依据文献方法[6-7],称取乙二氨四乙酸(EDTA)14.6 g,邻苯二胺质量 21.7 g(二者物质的量比 1∶4),然后加入乙二醇溶剂50~60 mL,160℃油浴反应约6 h,直到无水蒸气放出为止。缩合产物冷却至120℃倒入烧杯,边搅拌边加入200 mL蒸馏水。冷却至室温,变粘稠,加入适量无水乙醇,静置过夜。过滤得到紫红色粗产品,然后将粗产品用50 mL 95%乙醇重结晶2~3次得到粉红色固体产物,用少量丙酮洗涤,得到淡粉色固体粉末 21.9 g,产约 75.5%。
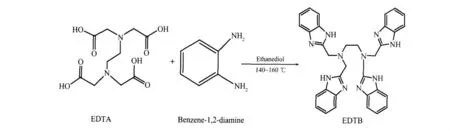
1.3 配合物的合成
称取提纯后的 EDTB(0.58 g,1 mmol)粉末溶解于20 mL甲醇与DMF混合液中,然后在60℃搅拌下加入 Mn(NO3)2·4H2O(0.18 g,1 mmol)水溶液,反应2 h,冷却过滤,得到淡黄色溶液。将溶液静置于室温下数周后析出浅棕色方形单晶。Anal.Calcd.for C37H39MnN13O7(%):C 53.33,N 21.81,H 4.62;Found(%):C 53.13,N 21.79,H 4.60。
配合物的紫外-可见吸收光谱显示在275 nm处有苯并咪唑环π-π*跃迁。配合物的红外吸收光谱是以KBr压片,在3800~3000 cm-1范围内强而宽的吸收峰带归属为芳环上νC-H和νN-H;2 920 cm-1附近的峰可归属为亚甲基(-CH2-)的振动吸收;1630~1655 cm-1归属为 νC=O;1620 及 1 450 cm-1附近两个吸收峰可以归属为苯环骨架振动吸收峰 (νC=H);745 cm-1归属为芳环上C-H的面外弯曲振动或NH的非平面振动;在远红外270~290 cm-1归属为烷氨氮与金属配位的νM=N特征振动峰。
1.4 晶体结构的测定和结构解析
选择尺寸大小为 0.30 mm×0.20 mm×0.20 mm 的浅棕色方形单晶,在室温295(2)K下,置于Brucker AXS SMART 1000 CCD单晶衍射仪上,用石墨单色器单色化的 Mo Kα 辐射(λ=0.71073)和 φ/ω 扫描方式最终的差值傅立叶图上,共收集到29628个衍射点,独立衍射点 8316 个,其中 6876 个(I>2σ(I))衍射点用于结构精修。晶体结构分析采用SHELXL程序完成,氢原子坐标通过理论加氢程序得到。所有氢原子均采用各向同性热参数,非氢原子采用各向异性热参数。对原子坐标和热参数用全矩阵最小二乘法进行了修正,并用PLATON程序绘出化合物的晶体结构图。配合物[Mn(EDTB)](NO3)2·DMF为正交晶系,P212121空间群,a=1.17252(6)nm,b=1.32083(6)nm,c=2.46191(12)nm,V=3.8128(3)nm3,Dc=1.451 g·cm-3,Z=4,F(000)=1732,Flack 指数为 0.02(15)。最终残差因子 R1=0.0522,wR2=0.1022。最终的差值傅立叶图上,最高电子密度峰为452 e·nm-3,最低电子密度峰为 -277 e·nm-3。晶体学数据和精修参数见表1,键长和键角见表2,氢键数据见表3。
CCDC:771022。

表1 配合物的晶体学数据和精修参数Table 1 Crystal data and structure refinement for the complex

表2 配合物[Mn(EDTB)](NO3)2·DMF部分的键长和键角Table 2 Selected bond lengths(nm)and angles(°)of[Mn(EDTB)](NO3)2·DMF

表3 [Mn(EDTB)](NO3)2·DMF的氢键数据Table 3 Hydrogen bond data for[Mn(EDTB)](NO3)2·DMF
1.5 配合物的PPO活性实验
分光光度法测定多酚氧化酶的活性,因操作简易、方便、快速,被广泛采用。PPO活性的测定可采用多种底物,主要有邻苯二酚、邻苯三酚、酪氨酸、氯原酸、多巴及多巴胺[8]。通过对比和筛选,本实验采用邻苯三酚为测试底物,实验结果较为敏感和稳定。
在 pH=8.2,Tris-HCl 5.0 mL 的缓冲液中分别加入双蒸水 4.7,4.5,4.3,3.9,3.7,3.2 mL 和模型配合物(30 μmol·L-1)0.0,0.2,0.4,0.6,0.8,1.0,1.5 mL,摇匀后在30℃水浴中恒温10 min,立即加入30℃恒温的邻苯三酚 0.3 mL,4.0 mmol·L-1并混匀,迅速转入1 cm石英比色皿中,1 min后开始测定,并每隔30 s分别记录体系在320 nm处的吸光度,共测定9~11个点。
在 5.0 mL,0.1 mol·L-1Tris-HCl的缓冲液 (pH=7.8,8.0,8.2)中分别加入 4.5 mL 双蒸水和 0.2 mL 模型配合物(浓度为 30 μmol·L-1),摇匀后在 30 ℃水浴中恒温10 min,立即加入30℃恒温的邻苯三酚0.30 mL(浓度为 4.0 mmol·L-1)混匀,迅速转入 1 cm石英比色皿中,立即开始测定,每隔30 s分别记录体系在320 nm处的吸光度,共测定9~11个点,然后每隔1 min测定1次,直到吸光度不再发生变化,并记录最大值。
2 结果与讨论
2.1 配合物的晶体结构
配合物中锰(Ⅱ)分别与来自EDTB分子的4个苯并咪唑氮原子(N(3),N(5),N(2),N(9))和 EDTB 分子的2个亚胺基氮原子(N(1),N(7))配位,形成1个六配位的扭曲八面体结构、DMF游离在外界。Mn2+离子周围的配位环境上 N(2),N(3),N(5),N(9)处于赤道位置,而N(1),N(7)处于轴向位置。标题配合物晶体的主要键长和键角列于表2,标题化合物的不对称单元椭球图见图1。从表2可知,亚胺基氮原子与 Mn2+之间的距离(Mn(1)-N(1)0.237 8(2)nm,Mn(1)-N(2)0.238 8(2)nm)长于苯并咪唑氮原子与Mn2+离子之间的距离(Mn(1)-N(3)0.216 1(2)nm,Mn(1)-N(5)0.2229(2)nm,Mn(1)-N(7)0.216 1(2)nm,Mn(1)-N(9)0.2228(2)nm),这与苯并咪唑失去 H+带负电荷的氮离子给电子的能力比亚胺基氮原子给电子能力强的事实相符合。标题配合物中处于轴向位置的 N(1)与处于赤道位置的 N(3),N(9),N(5),N(2)的键角大小分别为∠N(3)-Mn(1)-N(1)=75.71(8)°,∠N(9)-Mn(1)-N(1)=75.51(9)°,∠N(1)-Mn(1)-N(2)=76.68°,∠N(5)-Mn(1)-N(1)=89.61(8)°,前 3 个角偏离90°,而后1个角接近90°。处于轴向位置的N(7)与处于赤道位置的 N(1),N(2),N(9),N(3)的键角大小分别为∠N(7)-Mn(1)-N(5)=94.49(9)°,∠N(7)-Mn(1)-N(9)=92.60(9)°,∠N(7)-Mn(1)-N(2)=75.59(9)°,∠N(3)-Mn(1)-N(7)=134.01(9)°,前 2 个角接近 90°,而后 2 个角偏离90°。3对处于对角位置的氮原子键角大小为:∠N(7)-Mn(1)-N(1)=149.78(9)°;∠N(3)-Mn(1)-N(2)=148.79(8)°;∠N(9)-Mn(1)-N(5)=160.81(9)°,3 个角均偏离180°。标题配合物中心锰(Ⅱ)离子与与配合物[Mn(EDTB)Cl]Cl·H2O[8],[Mn(EDTB)(Ac)]Ac·EtOH[9],[Mn(EDTB)(PhCOO)]ClO4·EtOH·H2O[10]的中心金属锰(Ⅱ)离子的配位模式不同。这是因为配合物[Mn(EDTB)](NO3)2·DMF中心金属锰(Ⅱ)离子与来自EDTB分子的4个苯并咪唑氮原子和EDTB分子的2个亚胺基氮原子配位,形成1个六配位的扭曲八面体结构,而其他3个配合物中心金属锰(Ⅱ)离子只与配体EDTB分子的3个苯并咪唑氮原子和EDTB分子的2个亚胺基氮原子配位,同时与1个小分子配体配位形成六配位或者七配位的结构。另外,标题化合物中心锰离子的几何构型与近期报道的单晶 [Zn(EDTB)](NO3)2·C2H5OH[11],[Cu(EDTB)](NO3)2·C2H5OH[12],[Ni(EDTB)](NO3)2·C2H5OH[13]的中心离子的几何构型基本一致。标题化合物的单胞堆积结构中含有4个[Mn(EDTB)](NO3)2·DMF结构单元。阴离子与其邻近的配阳离子的苯并咪唑环上未与Mn(Ⅱ)配位的亚胺基氮形成氢键,与邻近的配阳离子EDTB的苯环上碳原子形成氢键。溶剂DMF分子也与其邻近的配阳离子EDTB的亚胺基相连的碳原子形成弱氢键。晶胞中的8个阴离子NO3-虽未参与配位,但参与了氢键的形成,同时起电荷平衡的作用。配合物具体氢键数据如表3所示,据此可知,整个晶体中除了范德华引力外,结构单元内部、结构单元之间还靠氢键来维系,分子间以较紧密的堆积方式彼此作用成一个整体。
2.2 配合物的PPO活性研究
在 30 ℃,0.1 mol·L-1的 Tris-HCl(pH=8.2)缓冲液中加入固定量的邻苯三酚溶液,先测定邻苯三酚自氧化速度,然后加入一定量的PPO模型配合物,测定邻苯三酚的自氧化速度。加入邻苯三酚1 min后开始记录,并每隔30 s分别记录体系在320 nm处的吸光度。
配合物-邻苯三酚反应体系中,自氧化时间t对吸光度A的线性回归方程:
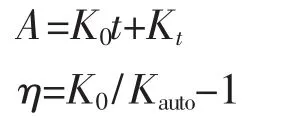
A为吸光度,t为自氧化时间,K0直线斜率,即邻苯三酚的自氧化速度,Kt为直线的截距。K0和Kauto分别代表体系中有或无PPO模型化合物时邻苯三酚的自氧化速率。邻苯三酚促进率为η,若改变加入PPO的浓度(c),可计算出不同浓度时的促进率,作出η-c曲线,再计算PC50(促进率为50%的浓度)。PC50越小,表明该PPO模型配合物活性越高[13]。反应体系的吸光度A对自氧化时间t的关系如图2,R2代表直线的拟合优度值。反应体系的回归参数及相应的促进率η见表4。从表4可以看出,在30℃,pH=8.2,配合物浓度在 0.1~10 μmol·L-1范围内,随着浓度的逐渐增大,配合物催化邻苯三酚的活性逐渐增大,促进率从51.5%增至72%。当浓度大于2 μmol·L-1时,促进率逐渐趋于稳定。经计算,可得该配合物 PC50约为 0.6 μmol·L-1。
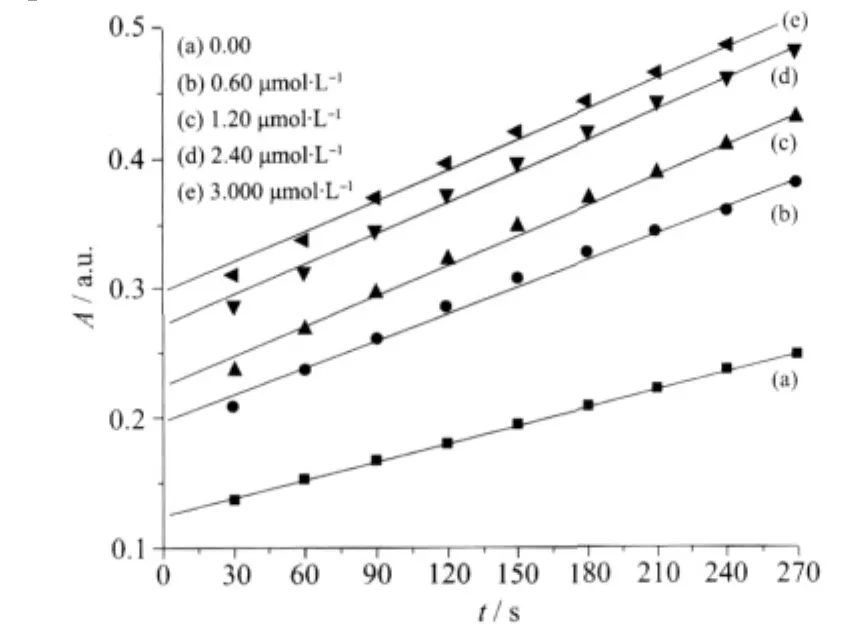
图2 Tris-HCl(pH=8.2)缓冲体系中吸光度随时间和配合物浓度变化关系Fig.2 Absorbance vs autoxidation time of pyrogallol in Tris-HCl(pH=8.2)buffer at a different concentration of the complex
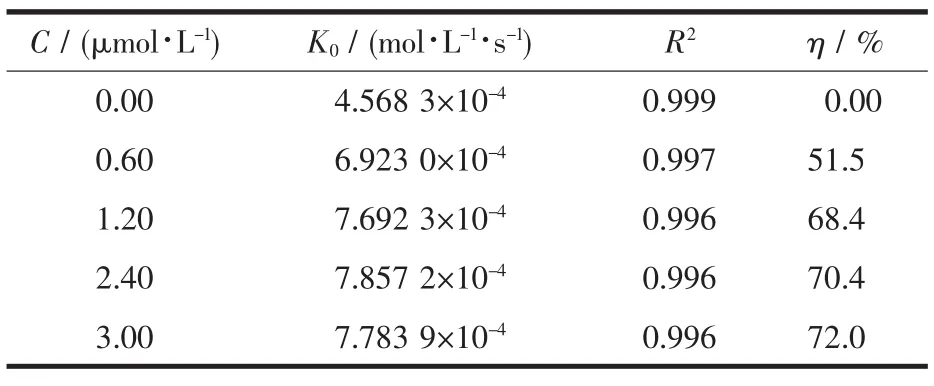
表4 配合物动力学参数及促进率ηTable 4 Kinetic data and η of the complex
在 30 ℃,底物邻苯三酚浓度为 4.0 mmol·L-1,模型配合物浓度为 6 μmol·L-1。采用 Tris-HCl,不同pH值的缓冲溶液中,配合物对邻苯三酚的催化氧化用动力学公式表述,配合物ln[(Af-Ai)/(Af-At)]~t关系图见图3,Af为反应完全时溶液的最终吸光度值,Ai为反应开始时溶液的吸光度值,At为反应中某时刻溶液的吸光度值。ln[(Af-Ai)/(Af-At)]~t关系图为直线关系,斜率为K说明此配合物的多酚氧化酶活性在上述条件下基本符合动力学一级反应。在pH=7.8~8.2 条件下,配合物的转化率 K 由 22.93 h-1增加到51.01 h-1(见表5),说明配合物的PPO活性随着pH值的升高而明显增强。综上可见,标题配合物的PPO模拟活性较高,而其模拟活性机理还有待进一步探讨。

图3 pH对PPO活性的影响Fig.3 Effect of pH on PPO activity

表5 pH条件对配合物的动力学数据的作用Table 5 Effect of pH on kinetic data of the complex
致谢:感谢华中师范大学化学院刘长林教授悉心的指导和修改!
[1]Qiu J H,Liao Z R,Meng X G,et al.Polyhedron,2005,24:1617-1623
[2]Klabunde T,Eicken C,Sacchettini J C,et al.Nat.Struct.Biol.,1998,5:1084-1090
[3]LI Zhong-Guang(李忠光),GONG Ming(龚明).Journal of Yunnan Normal University(Yunnan Shifan Daxue Xuebao),2005,25:44-49
[4]DAI Ya(戴亚),SHI Chun-Hua(施春华),TANG Hong(唐宏),Acta Tabacaria Sinica(Zhongguo Yancao Xuebao),2001,7(4):7-12
[5]HUANG Jian-Shao(黄建韶),ZHANG Hong(张洪),TIAN Hong-Xian(田红现).Food and Machinery(Shipin Yu Jixie),2001,83:21-22
[6]Qin S D,Feng S S,Zhang H M,et al.Acta Crystallogr.,2004,E60:o1121-o1122
[7]Hendriks H M J,Birker P J M W L,van Rijn J.J.Am.Chem.Soc.,1982,104:3607-3617
[8]WU Hong-Mei(吴红梅),XIAO Hui(萧慧),LI Gang(刘刚),et al.Journal of Tea Business(Chaye Tongbao),2004,26(2):62-64
[9]Li Q X,Luo Q H,Li Y Z,et al.Eur.J.Inorg.Chem.,2004:4447-4456
[10]Liao Z R,Zheng X F,Luo B S,et al.Polyhedron,2001,20:2813-2821
[11]SunY,Ying Y X,Chen K,et al.Pharmacol.Res.Commun.,2005,25:10-11
[12]Liu Y C,Ma J F,Hu N H,et al.Acta Crystallogr.,2003,E59:m361-m363
[13]Chen Z F,Liao Z R,Li D F,et al.J.Inorg.Biochem.,2004,98:1315-1318
[14]QIN Shi-Dong(覃事栋),FENG Si-Si(冯思思),ZHANG Hong-Mei(张红梅),et al.Acta Chimica Sinica(Huaxue Xuebao),2005,63(13):1155-1160
Synthesis and Crystal Structure of Complex[Mn(EDTB)](NO3)2·DMF and Its Polyphenol Oxidase Activity
WANG Li*,1WEI Li-Mei1ZHANG Yong1,2PEI Ya-Mei1
(1School of Chemistry,Central China Normal University,Wuhan 430079,China)(2School of Chemical and Materials Engineering,Huangshi Institute of Technology,Huangshi,Hubei 435003,China)
The title complex[Mn(EDTB)](NO3)2·DMF[EDTB=N,N,N′,N′-tetrakis-[(2-benzimidazolyl)methyl]-1,2-ethanediamine]has been synthesized and characterized by elemental analysis and single crystal X-ray diffraction method.The crystal of mononuclear Mn(Ⅱ) complex belongs to orthorhombic crystal system with space group,P212121,a=1.172 52(6)nm,b=1.320 83(6)nm,c=2.461 91(12)nm,V=3.812 8(3)nm3,Dc=1.451 Mg·m-3,Z=4,F(000)=1 732.The final R(I>2σ(I)):R1=0.052 2,wR2=0.102 2 and R(all data):R1=0.065 2,wR2=0.107 5,Flack parameter:0.02(15).The manganese(Ⅱ) atom of the cation has distorted octahedral coordination geometry and is six-coordinated by four nitrogen atoms of the benzimidazole groups and two tertiary amine nitrogen atoms from the ligand.Polyphenol oxidase activity toward pyrogallol has been studied spectroscop ically.In the 0.1 ~10 μmol·L-1rang,promotion rates of the complex increase from 51.5%to 82.2% .The turnover numbers of the complex toward pyrogallol increase from 22.93 h-1to 51.01 h-1at 30 ℃,showing polyphenol oxidase activity of the complex increasing with pH.CCDC:771022.
manganese(Ⅱ)complex;EDTB;crystal structure;polyphenol oxidase activity
O614.7+11
:A
:1001-4861(2011)01-0125-06
2010-06-27。收修改稿日期:2010-10-12。
国家自然科学基金(No.21001047)和深圳微生物基因工程重点实验室开放基金资助项目。
*通讯联系人。E-mail:wl_928@mail.ccnu.edu.cn;会员登记号:S060015331M。
