高效液相色谱法测定水产品中呋喃西林的研究
2011-10-24岑剑伟李来好杨贤庆郝淑贤辛少平周婉君
岑剑伟,李来好,杨贤庆,郝淑贤,魏 涯,辛少平,石 红,周婉君
(中国水产科学研究院南海水产研究所,广东广州 510300)
高效液相色谱法测定水产品中呋喃西林的研究
岑剑伟,李来好*,杨贤庆,郝淑贤,魏 涯,辛少平,石 红,周婉君
(中国水产科学研究院南海水产研究所,广东广州 510300)
建立了高效液相色谱测定水产品中呋喃西林残留量的方法。通过对比不同有机溶剂的提取效果,表明使用单溶剂提取回收率均不高,采用丙酮和二氯甲烷的混合溶液提取效果更好,其中比例为3∶7时最佳。比较了振荡法、超声波法和超声波加热法等的提取能力,结果表明,超声波加热法提取效果最好。以丙酮-二氯甲烷作为提取剂、超声波加热(35kHz,强度100%,40℃)进行提取,提取时间15min、提取2次,提取率可达90%以上,方法回收率在75%~100%之间,相对标准偏差小于10%,检测限能达到0.5μg/kg。本方法操作简便,回收率较高,稳定性好。
呋喃西林,超声波提取,高效液相色谱
呋喃西林(Nitrofurazone)是一种具有5-硝基呋喃基本结构的广谱抗菌药,主要通过干扰细菌体内的氧化还原酶系统,曾经被广泛应用于家禽、家畜、水产养殖动物传染病的预防和治疗,也被作为饲料药物添加剂。上世纪90年代被发现具有致畸、致突变和致癌作用,由于其代谢产物始终存在,严重危害人体健康,世界各国均严禁在治疗和饲料中使用。欧盟在1995年就规定禁止在食物中使用该类药物,并于2003年确定水产品中硝基呋喃类药物及其代谢物的检测限为1μg/kg[1]。我国也于2002年开始禁止在所有食品动物中使用硝基呋喃类抗生素。目前,对于食品中呋喃西林的检测方法,大多是针对呋喃西林代谢物的检测方法[2-4]。研究表明,在食品、环境中氨基脲(SEM)并不是单一由呋喃西林代谢而来,还可能是因为包装或加工过程中其他污染源造成的,欧盟食品委员会也对SEM的毒性重新进行了界定,目前不再将SEM作为判断呋喃西林污染的唯一标准[5]。通过检测氨基脲含量来代表呋喃西林的使用情况并不是很准确,因而有必要建立关于呋喃西林原药的检测方法,以便更好地发现和监管呋喃西林在水产养殖中非法使用的情况。呋喃西林检测方法主要有免疫检测法和理化检测法两类,免疫检测法特异性好、灵敏度高、精密度高,样品前处理简单,操作简便,不需要大型仪器设备,但其经常会出现假阳性结果[6-7];理化检测方法有紫外分光光度法、薄层色谱法、高效液相色谱法、液质联用技术等[6]。紫外分光光度法、薄层色谱法由于灵敏度低已逐渐淘汰,高效液相色谱法虽然灵敏度能够达到检测要求但往往样品处理过程复杂,液质联用法测定则所需设备昂贵,检测成本较高,同时也存在样品处理过程复杂等的缺点[8]。本文重点研究水产样品前处理方法,提升样品的呋喃西林的提取率,利用高效液相色谱检测,建立快速、简便、准确、灵敏度高的水产品中呋喃西林残留检测方法。
1 材料与方法
1.1 材料与仪器
罗非鱼、南美白对虾 市售;呋喃西林标准品Dr.Ehrenstorfer公司;二氯甲烷、甲醇、乙腈 色谱纯,德国默克;其他试剂 均为分析纯;实验用水 符合GB/T6682一级水标准。
AgiLent1100高效液相色谱 美国安捷伦公司; N-1000旋转蒸发仪 EYELA日本东京理化;TDZ5-WS自动平衡离心机 湖南湘仪仪器公司;Transonic TI-H10超声波清洗仪 强度100%,德国 Elma; N-EVAPTM112氮吹仪 美国Organomation;GB204分析天平 瑞士梅特勒。
1.2 实验方法
1.2.1 试剂配制 丙酮-二氯甲烷提取液:取丙酮与二氯甲烷按体积比(3∶7)混合;标准储备溶液: 100μg/mL,称取呋喃西林标准品10.0mg,加入乙腈溶解,定容到100mL,4℃贮存;标准工作液:取标准贮备液,加50%乙腈水溶液稀释配制成浓度为0.005、0.01、0.02、0.05、0.1、0.2、0.5、1、2μg/mL的梯度系列。
1.2.2 样品前处理 将样品搅碎或均质,称取10.00g于100mL离心管中,加提取液20mL,再加入无水硫酸钠(10g),充分摇匀,按下文研究方法提取后,4500r/min离心5min,取上清液,沉淀再加入20mL提取液重复提取1次。合并上清液于蒸馏瓶中,在旋转蒸发仪45℃水浴减压蒸馏至近干,加入2mL 50%乙腈水溶液洗瓶,取洗液过0.22μm滤膜,备用。
1.2.3 色谱条件 色谱柱:Agilent Zorbax SB-C18,250mm×4.6mm(5μm);流动相A:乙腈;流动相B:超纯水;进样量:20μL;流速:1.0mL/min;柱温:30℃;检测信号:365nm。使用梯度洗脱程序如表1所示。

表1 梯度洗脱程序
1.2.4 计算公式 样品中呋喃西林含量计算公式为:
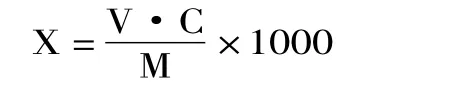
式中:X-样品中呋喃西林的含量,μg/kg;C-提取液中呋喃西林的含量,μg/mL;V-样品液最终定容体积,mL,本方法为2mL;M-样品的称取量,g。
2 结果与分析
2.1 提取液的选择
实验中分别使用二氯甲烷、丙酮、甲醇、乙腈和乙酸乙酯等溶剂提取鱼肉组织,发现回收率均较低(小于60%)。二氯甲烷、乙酸乙酯提取时样品液较为干净,而用乙腈、丙酮或甲醇提取使得提取液中含大量蛋白等杂质,在后续蒸馏步骤中容易引起爆沸。另外,乙酸乙酯、乙腈蒸馏时所需温度较高,长时间高温处理会导致呋喃西林降解,引起实验误差。有文献报道,使用丙酮与二氯甲烷的混合溶液提取,可以提高回收率[10]。对比不同比例的丙酮和二氯甲烷混合液的提取效果,结果如表2所示。单独使用二氯甲烷或纯丙酮提取回收率很低,但丙酮与二氯甲烷按一定比例混合提取时,回收率有较明显的提高,当丙酮比例在20%~50%之间时,回收率达到85%以上,且较稳定。另外,从回收率角度看,丙酮比例在20%附近时回收率波动比较大,而在加热提取过程中溶剂的蒸发不可避免,对提取的稳定性会造成一定的影响。30%附近时则变化比较平缓,溶剂蒸发对之影响较小,故选取丙酮与二氯甲烷(3∶7)混合液作为提取液。
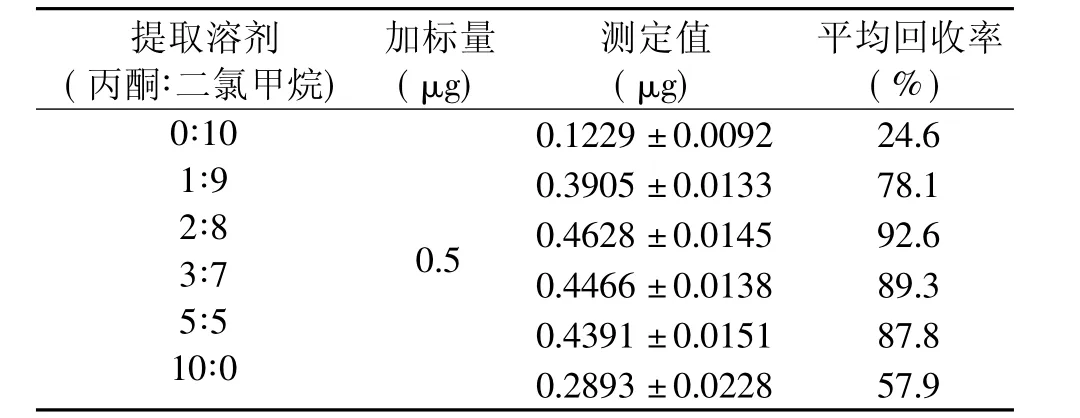
表2 不同比例混合提取液提取效果比较
2.2 提取方法的研究
2.2.1 提取方法比较 将空白鱼肉与呋喃西林标样充分混合,称取10.00g加标样品,加入20mL丙酮∶二氯甲烷(3∶7)提取液,分别用振荡(230r/min)、超声波(35kHz,强度100%)、超声波加热(35kHz,强度100%,40℃)三种方法提取一次,处理不同时间,浓缩提取液,测定提取总量,如表3所示结果,超声波提取比振荡提取的能力强,超声波加热辅助提取效果更好,15min之后提取量基本保持稳定。2.2.2 超声波提取率 将空白鱼肉搅碎加入呋喃西林充分混合,放置一段时间,称取10.0g加标样品,加入20mL丙酮∶二氯甲烷(3∶7)提取液,于超声波加热(35kHz,强度100%,40℃)提取15min,提取4次,分别测定每次提取液中所提取出呋喃西林的量,做4个平行。结果如表4所示,在设定条件下,提取1次提取率接近70%,提取2次后样品中呋喃西林含量已经很低,前两次提取量占总提取量的90%以上。重复提取两次已经能满足分析的要求,因此对样品处理时超声波提取两次即可。
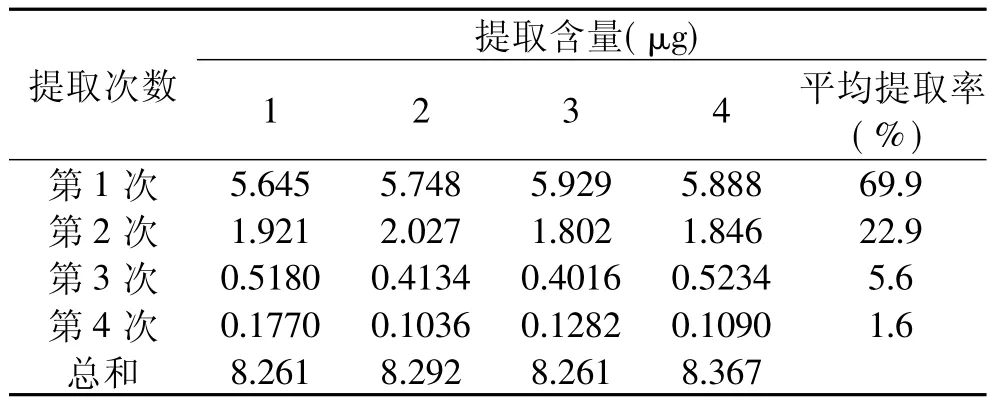
表4 超声波热辅助提取
2.3 方法评价
2.3.1 方法线性范围和检测限 配制呋喃西林标准工作液,用液相色谱分析,测定不同浓度工作液的峰面积,并做峰面积-浓度校正曲线,获得曲线方程为: Area=96.409Amount-0.00034,相关系数为:0.99998。呋喃西林在0.005~2μg/mL范围内线性良好。校正曲线图与标准色谱分别见图1、图2。
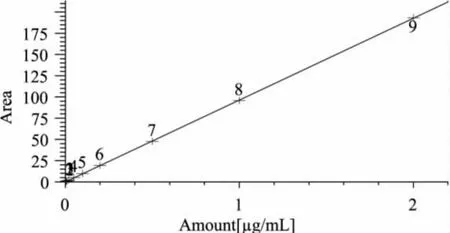
图1 校正曲线图
以0.005μg/mL浓度的呋喃西林溶液进样测定,计算得对应信噪比(S/N)为12,将浓度降低1倍进样分析,色谱峰较小,S/N值小于3。因而拟定以检测方法的标准曲线最低点方法为该法的检测限为0.005μg/mL,换算为水产样品对应值为0.5μg/kg,与本方法定量下限值相同。
2.3.2 准确度和精密度 称取10g搅碎样品,分别加入不同浓度标准品,以丙酮-二氯甲烷为提取液,超声加热提取,按以上最佳条件操作测定。另外做一份空白对照,实验结果见表5。本方法回收率在75%~100%之间,相对标准偏差小于10%,方法回收率高、稳定性好。按本方法提取,鱼肉和虾肉的样品色谱图均较干净,鱼肉样品色谱图杂质峰比虾肉样品更少。标准色谱图、虾空白样品及加标样品见图2。
3 讨论
3.1 提取液
本文比较了二氯甲烷、丙酮、甲醇、乙腈和乙酸乙酯等有机溶剂提取水产品中呋喃西林的效果,发现单溶剂的提取效果均不好,回收率较低。但使用丙酮和二氯甲烷混合溶剂提取时,回收率显著提高,丙酮比例在20%~50%之间的提取效果均较好,回收率高且较稳定。吴富忠等认为丙酮比例超过20%时提取液中部分水不能脱去而影响测定结果[10],但按照本文操作,提取时即加入足量无水硫酸钠脱水不存在类似问题。本文中使用丙酮和二氯甲烷混合液提取,提取液较干净,因而可以直接进样,无需再做进一步的纯化和净化,简化了操作。
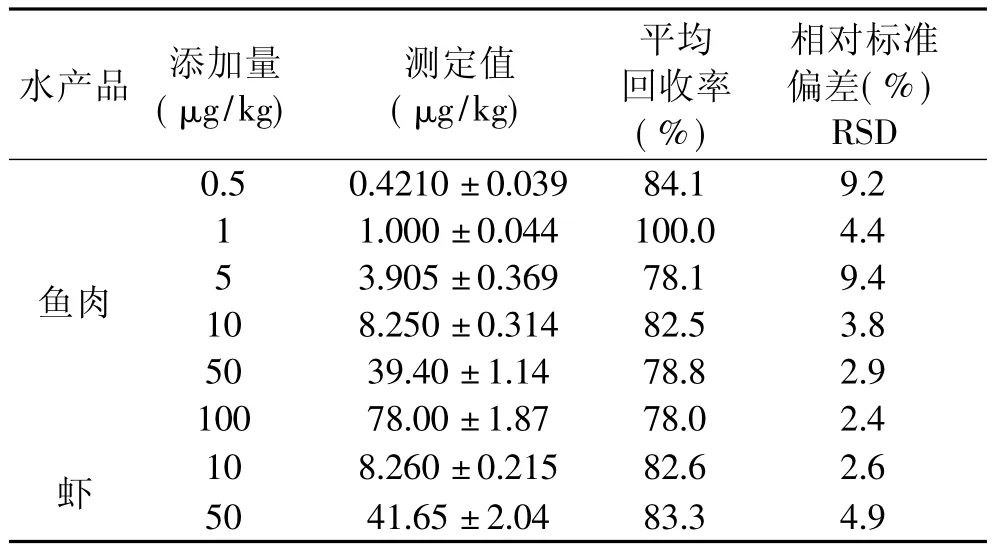
表5 方法回收率
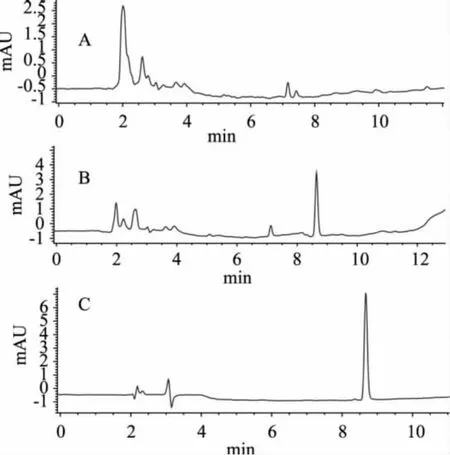
图2 色谱图
3.2 提取方法
以振荡、超声波、超声波热辅助等提取方法做比较,发现超声波与振荡相比较,超声波作用更有利于呋喃西林从肉组织中溶解出来,前者的提取能力更强。超声波再辅以加热进行提取,提取效率更高,可有效缩短操作时间,但仍需作用15~20min才能达到峰值。本文以超声波辅以加热的方法提取,提取15min,提取两次可将样品中90%以上的呋喃西林提取出来。但在测定方法回收率时,往往会低于此值,分析其可能原因是呋喃西林与生物样品接触后,在酶的作用下部分被降解,导致检测结果偏低甚至出现假阴性。因此,在操作过程中样品尽量避免长时间置于常温下,不能及时处理的样品应在低温下冻存。
本文研究了水产样品中呋喃西林残留量的提取方法,以丙酮-二氯甲烷作为提取液,超声波辅助加热法提取,提取效果最好,样品提取液杂质少,不需要后续净化步骤;方法回收率在75%~100%之间,稳定性好,相对标准偏差小于10%,检测限能达到0.5μg/kg。与超高效液相色谱串联质谱法(0.3μg/kg)接近[13],但样品处理过程中不需要使用固相萃取等步骤,操作更为简化。该法除了可以应用于水产品中呋喃西林含量的检测,同样也可作为饲料中呋喃西林检测方法的参考。
[1]田蕴,罗晓琴.水产品硝基呋喃类药物残留检测[J].动物保健,2006(7):45-47.
[2]DB33/T599-2006,水产品中硝基呋喃类代谢物残留量的测定-液相色谱-串联质谱法[S].浙江省地方标准,2006.
[3]GB/T18932.24-2005,蜂蜜中呋喃它酮、呋喃西林、呋喃妥因和呋喃唑酮代谢物残留量的测定方法液相色谱-串联质谱法[S].北京:中国标准出版社,2005.
[4]Jane Kelly Finzi,Jose Luiz Donato,Mauro Sucupira,et al. Determination of nitrofuran metabolites in poultry muscle and eggs by liquid chromatography-tandem mass spectrometry[J].Journal of Chromatography B,2005,824(1/2):30-35.
[5]李春风,康海宁,岳振峰,等.食品中氨基脲来源的研究进展[J].中国兽医杂志,2010,46(2):88-89.
[6]何方洋,沈建忠,万宇平,等.呋喃西林及其代谢物残留检测研究进展[J].中国兽药杂志,2009,43(4):51-54.
[7]蔡宝亮,余兵,陶宏锦,等.单克隆抗体介导的呋喃西林残留检测方法及其应用:中国,200610041481.6[P].2006-09-28.
[8]徐一平,胥传来.动物源食品中硝基呋喃类物质及其代谢物残留的检测技术研究[J].食品科学,2007,28(10):590-593.
[9]Jorge Barbosa,Sara Moura,Rita Barbosa,et al.Determination of nitrofurans in animal feeds by liquid chromatography-UV photodiode array detection and liquid chromatography-ionspray tandem mass spectrometry[J].Analytica Chimica Acta,2007,586:359-365.
[10]吴富忠,欧阳立群.水产品中呋喃唑酮、呋喃西林药物残留的HPLC法测定[J].中国卫生检验杂志,2006,16(7): 812-813.
[11]李佐卿,殷居易,倪梅林,等.高效液相色谱法检测水产品中硝基呋喃类药物残留[C].第十五次全国色谱学术报告会文集(下册),2005:440-441.
[12]葛宝坤,王云凤,贺信.高效液相色谱法测定鸡肉、水产品中呋喃西林和呋喃唑酮残留量的研究[J].中国卫生检验杂志,2002,12(6):661-662.
[13]徐英江,任传博,田秀慧,等.海产品中硝基呋喃类原药的超高效液相色谱串联质谱测定[J].分析测试学报,2010,29 (4):327-330.
Determination of nitrofuran residues in aquatic products by HPLC method
CEN Jian-wei,LI Lai-hao*,YANG Xian-qing,HAO Shu-xian,WEI Ya,XIN Shao-ping,SHI Hong,ZHOU Wan-jun
(South China Sea Fisheries Research Institute,Chinese Academy of Fishery Sciences,Guangzhou 510300,China)
A High-performance liquid chromatography(HPLC)method for the nitrofuran residues in aquatic products was proposed.Extracted by methylene chloride,acetone,methanol,acetonitrile and ethyl acetate respectively,the recovery rate of nitrofuran cannot meet the requirement.Better result could be acquired by extracting with a mixed solution of acetone and dichloromethane,while the ratio was 3∶7.Ultrasonication assisted by heating was the best method for facilitating the extraction,comparing with oscillation,ultrasonication methods. Extracted with Acetone-dichloromethan agent,ultrasonication assisted by heating(35kHz,intensity 100%,40℃),treated twice with 15min each time,the nitrofuran can be extracted well,the extraction rate was higher than 90%.A method,of simplicity,high recovery and good stability,which the recovery was 75%~100%,the relative standard deviation was less than 10%,and the detection limit can reach 0.5μg/kg was attained.
nitrofurazone;ultrasonic extraction;high-performance liquid chromatography
TS207.3
A
1002-0306(2011)12-0455-04
2011-08-30 *通讯联系人
岑剑伟(1976-),男,研究生,主要从事水产品加工与质量安全研究。
国家农业产业技术体系项目(CARS-49);国家农业科技成果转化资金项目(2010GB23260577,2009GB2E200303,2010GB2E000335); 广 东 省 科 技 计 划 项 目(2009A020700004,2008A020100006,2009B020201003);广东省海洋渔业科技推广项目(A200899B02,A200901C01);农业部中央级公益性科研院所基本科研项目(2010YD07)。
