RTH 结构分子筛酸性的密度泛函理论研究
2011-10-12彭西来桂建舟江海英高玉环
彭西来, 桂建舟, 刘 丹, 江海英, 杨 爽, 高玉环
(辽宁石油化工大学化学与材料科学学院,辽宁抚顺113001)
RTH 结构分子筛酸性的密度泛函理论研究
彭西来, 桂建舟, 刘 丹*, 江海英, 杨 爽, 高玉环
(辽宁石油化工大学化学与材料科学学院,辽宁抚顺113001)
采用密度泛函理论中的广义梯度近似方法(GGA)对RTH结构分子筛的酸性进行了研究。考察了4个不同T位的A l的替代能以及16个不同Brönsted酸位的脱质子能,从而推断可能的Al的取代位以及不同酸位的酸性强弱。结果表明,RTH结构分子筛上最有可能的A l的取代位为T4,位于八元环的孔口(0.56 nm×0.25 nm);从结构和酸性考虑,易与反应物接触且酸性较强的Brönsted酸位点在A l4-O9(H)-Si2和A l4-O16(H)-Si4上,并且这两个酸位点都指向两个八元环孔口。
RTH; 酸性; 分子筛; 密度泛函理论
近年来,CHA类型的SSZ-13和SAPO-34分子筛在M TO(M ethanol-to-Olefin)反应中表现出优越的催化活性,受到广泛关注。但是SAPO-34分子筛催化M TO反应时,一个显著的特征是催化剂易于积炭失活[1]。RTH分子筛是1995年首次合成的二维八元环分子筛,它是由八元环组成的平行于a轴和c轴二维孔道,其孔道大小分别为0.41 nm×0.38 nm和0.56 nm×0.25 nm[2]。自发现以来,由于它具有独特的结构特征,这种沸石已经被预期在催化方面和吸附领域呈现独一无二的优越性。Toshiyuki Y等[2]最近报道了无模板剂法合成RUB-13并应用于M TO的反应中,与传统的SAPO-34分子筛相比,它对丙烯有更好的选择性。
RTH的单晶胞中含有96个原子,它存在4个结晶学上不同的T位(即硅氧或铝氧四面体):T1,T2位于0.41 nm×0.38 nm的八元环孔口,T2,T4位于0.56 nm×0.25 nm的孔口处,T3位于RTH笼的内壁(如图1所示)。尽管实验上可以利用固体核磁共振或红外光谱来检测Si和A l原子的分布,但是仍无法准确定位。实验方法的局限性及铝分布问题的重要性为化学理论计算的应用提供了充分的机会,而化学理论计算在研究各个独立T位置被范区A l原子取代后的相对能量,酸性强度以及稳定性是很有用的,同时它还可以确定分子筛中B酸性点的可能位置[3-4]。因此研究人员一直在试图计算各个独立T位置被铝原子取代后的相对能量。由于A l原子取代Si原子以后,将影响其周围骨架的结构和电子环境,因此应用化学计算可以预测骨架A l原子的位置和分子筛酸性。M TO反应是一个酸催化反应,其酸位是催化反应的中心,要进一步从分子水平上了解该分子筛的催化机理,需要对其不同酸位进行研究。
本课题组曾多次采用密度泛函理论(DFT)方法研究分子筛催化剂的结构和反应物在分子筛表面的吸附行为[5-7]。在此研究工作基础上,本文通过分子模拟的方法详细研究了RTH分子筛的结构和不同T位上形成B酸位的难易程度及其可能形成的16个B酸位上的酸性强弱顺序,为研究该类分子筛上的酸催化反应提供理论基础。
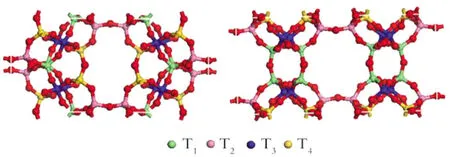
Fig.1 Labelling of four different T sites in the unit cell of RTH zeolite图1 RTH单晶胞中不同T位的标注
1 模拟建立和计算方法
分子筛模型的选取对计算结果至关重要,需充分考虑计算精度和计算时间的需要,为此实验选取的模型含有3层骨架原子,包含5个T原子并且以A l原子为中心,即A l-[OSi(OSiH3)4]4(T=1,2,3,4),其结构如图2所示。图中T原子为1~4位的不同骨架Si所在的位置,最外层的骨架O原子去掉后形成的悬断键用H原子饱和。由于对模拟分子筛结构的簇模型完全优化会导致得到的构型不一定符合所研究的分子筛,因此本文只进行了局部优化,即只计算中心原子和相邻的4个O原子,其它骨架原子固定在原来晶体结构的坐标上。这样的优化过程既保证了A l原子周围的骨架结构,又使整个模型保持RTH分子筛的骨架结构。当中心原子为Si原子时,整个体系为电中性,用A l原子替代后整个体系加一个负电荷。
理论计算平台是美国Accrlrys公司开发的Materials Studio 3.1软件包中的Dmol3模块[8]。计算参数设置如下:计算方法为广义梯度近似(GGA),选择BL YP泛函来处理交互相关能,基组采用可极化的双数基组(DNP),电子的处理采用所有电子参与计算的方法,系统自旋状态为Spinrestricted,计算采用非对称性,自洽场(SCF)参数的建立使总能量收敛至1×10-5Ha(1 Ha≈2 625.5 kJ/mol)。
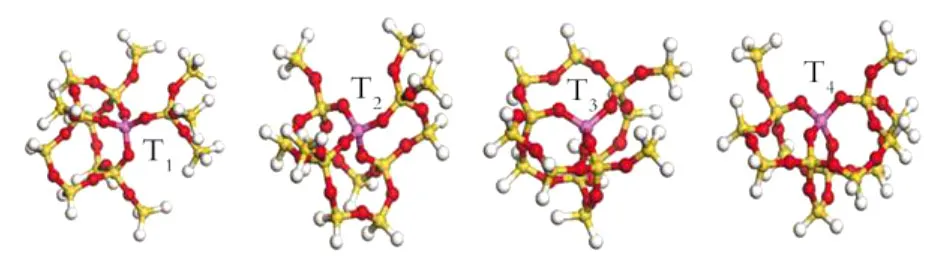
Fig.2 Selected clusters for different T sites of RTH zeolite after Al substitution图2 不同T位的A l取代后的模型搭建
2 结果与讨论
2.1 分子筛A l替代能
表1为4个不同T位的A l取代前后的中心团簇能量。由表1可知,原始的分子筛模型在优化前后能量基本吻合。A l替代之后,各T位的能量均相应增加,说明A l原子较难替代Si进入RTH的骨架结构,这与实际上只有同时存在B元素的情况下,A l原子才容易被引入到分子筛中的实验结果是相吻合的。上述结果说明各T位的骨架结构稳定性是存在差异的,其中T2位的能量总是明显高于其它位点,说明该T2位的晶格能最高;而T1位的能量总是明显低于其他位点,说明该T1位的晶格能最低。
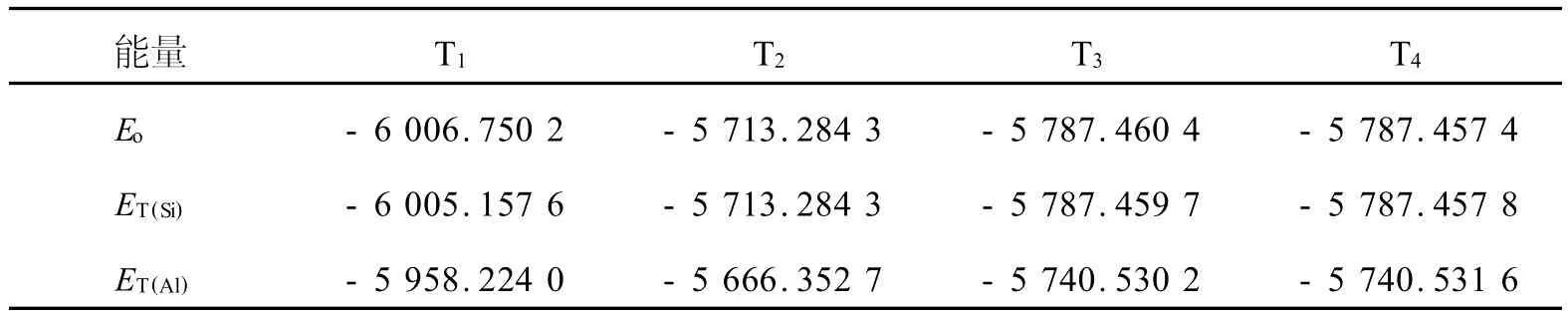
表1 A l取代4个不同T位前后的团簇的能量Table 1 Total energy of clusters of four different T sites before and after Al substituation Ha
RTH结构分子筛A l替代能可用公式(1)表示:
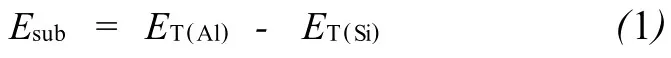
由公式(1)可知,相对取代能越高,说明该T位A l取代Si的难度越大。
图3为不同T位上的A l的相对取代能,通过分析各位点T中心原子Si被A l取代之后的总能量差,可知A l在不同T位上的相对取代能的顺序为T4<T3<T2<T1,其中各不同位点的替代能之间的最大差别为7.94×10-3Ha。A l原子将优先位于替代能小的位点T4上,T3,T2和T1的替代能比T4分别高2.35×10-3,5.46×10-3,7.94×10-3Ha。
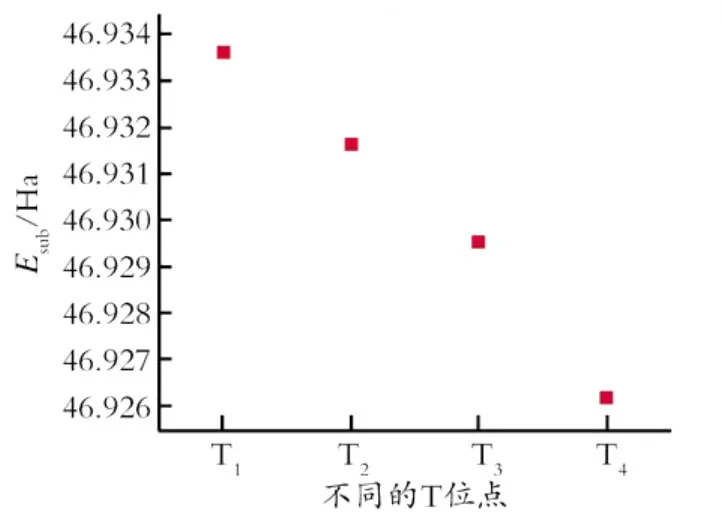
Fig.3 Relative Al substitution energy on four different T sites图3 不同T位上的A l的相对取代能
B rönsted酸的分布信息主要是从两个方面来考虑:一是A l取代特定T位时其质子的稳定性,二是A l在不同T位上的相对稳定性。当分子筛骨架上正四价的Si原子被正三价的A l原子取代时,形成正四面体配位的铝就引入了一个负电荷(A lO-4),为了弥补这个负电荷就必须引进一个质子。每个A l原子周围有4个骨架氧,任何一个都可以直接连接这个质子,理论上RTH结构分子筛中存在16种可能的构型。这时RTH分子筛A l替代能可以用公式(2)表示:
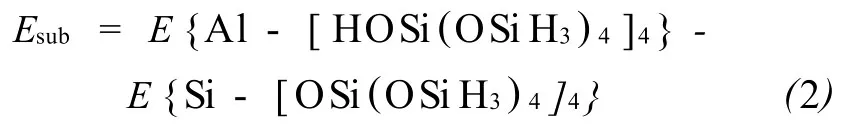
分析计算结果可知,各T位A l的替代能大致呈如下趋势:T4<T3<T2<T1。对于每个T位,直接连接质子的骨架氧不同,其得到的酸性中心的A l的替代能也有很大差异。其中从能量角度,比较易于形成的Brönsted酸位点应该在A l2-O8(H)-Si1,A l1-O6(H)-Si1,A l2-O9(H)-Si4,A l3-O5(H)-Si1,A l4-O9(H)-Si2和A l4-O16(H)-Si4(如表2所示)。其中A l2-O8(H)-Si1,A l1-O6(H)-Si1,A l2-O9(H)-Si4,A l4-O9(H)-Si2和A l4-O16(H)-Si4都指向两个八元环孔口,易与反应物相互作用。
2.2 分子筛脱质子能
表2为每个不同的B酸位(T(A l)H/T(Si))上的A l的替代能。

表2 不同的B酸位上的A l的替代能Table 2 Al substitution energy on different acid sites Ha
除了计算RTH分子筛不同T位的A l原子的取代能,还系统研究了不同A l取代位置的酸性的强弱。许多分子筛固体酸的酸性可以用脱质子能来表示。脱质子能定义为从质子化的模型中将质子移至无限远处所需的能量,即:
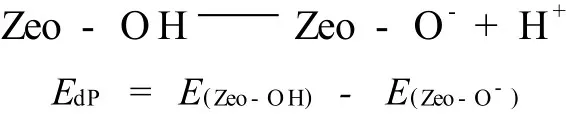
其中,E(Zeo-OH)和E(Zeo-O-)分别为分子筛质子化模型和去质子模型的总能量。尽管脱质子能在实验上很难直接测得,但在理论上常用来表征质子酸的相对强弱,它很容易通过量子化学计算得到。脱质子能越小,对应的酸性越强;相反,脱质子能越大,对应的酸性越弱。
表3给出了RTH分子筛上4个不同T位上的脱质子能。对于每个T位,直接连接质子的骨架氧不同,其得到的脱质子能也有很大差异。分析计算结果可知,A l替代后模型从能量角度越稳定,则其脱质子能越高。对于T1,T2来说,脱质子能较低的构型是A l1-O7(H)-Si3和A l2-O10(H)-Si2,但它们的A l替代能很高(见表2)。由于预测的是A l原子的有可能的取代位(即有可能的Brönsted酸落位)是T4和T3,所以应首先考虑的A l替代能较低的那些位置T3,T4位的酸度变化。计算结果表明这些位置的脱质子能的顺序如下:A l4-O15(H)-Si3>A l3-O15(H)-Si4>A l3-O7(H)-Si1>A l3-O13(H)-Si4>A l4-O13(H)-Si3>A l4-O9(H)-Si2>A l4-O16(H)-Si4>A l3-O5(H)-Si1。考虑到H质子的指向和与反应物接触的难易,易于取代且易与反应物作用的B酸位有A l4-O16(H)-Si4和A l4-O9(H)-Si2。
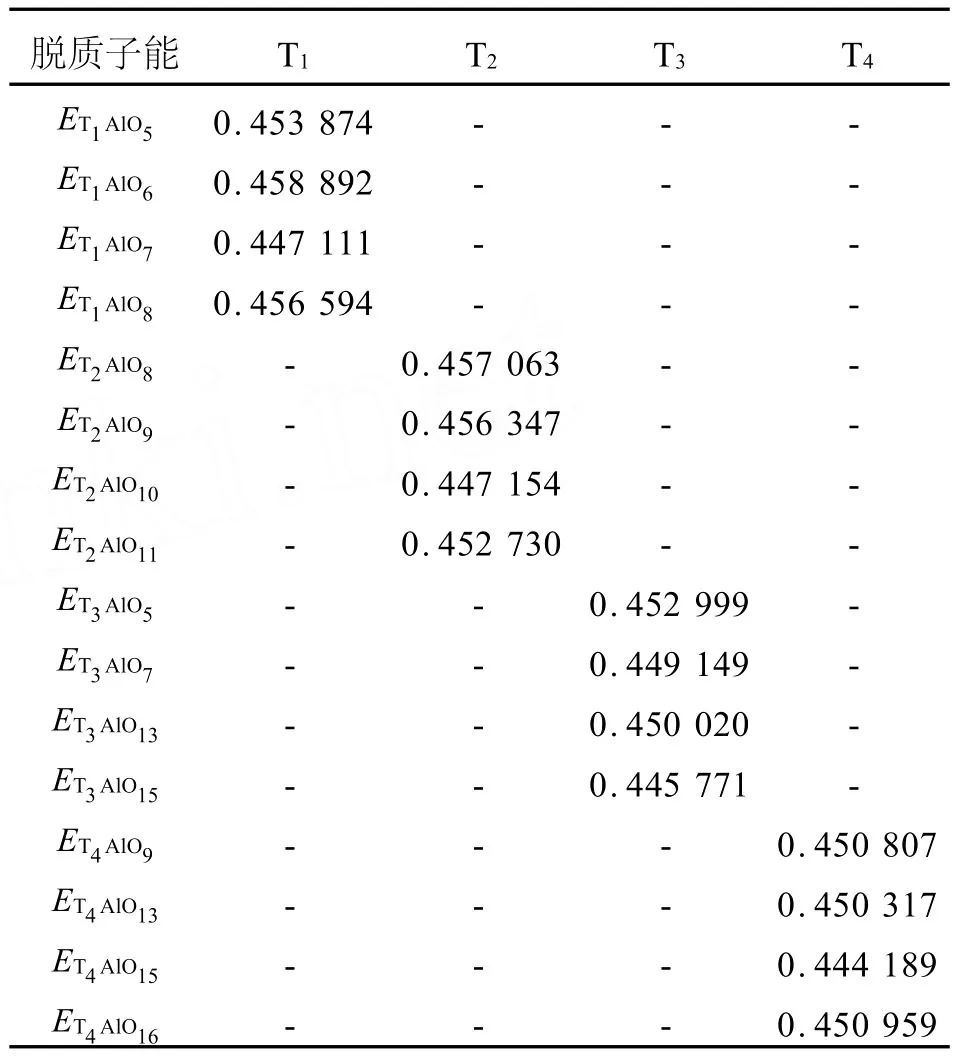
表3 RTH分子筛上不同T位上的脱质子能Table 3 Deprotonation energies of different Brönsted acid sites Ha
[1]刘红星,谢在库,张成芳,等.甲醇制烯烃(M TO)研究新进展[J].天然气化工,2002,27(3):49-53.
[2]Toshiyuki Y,Masato Y,Hiroyuki I,et al.Diversification of RTH-type zeolite and its catalytic app lication[J].Angew.chem.int.ed.,2009,48(52):9884-9887.
[3]Brand H V,Curtiss L A,Iton L E.Computational studies of acid sites in ZSM 5:dependence on cluster size[J].J.phys.chem.,1992,96(19):7725-7732.
[4]A lvarado-Swaisgood A E,Barr M K,Hay P J,et al.Abinitio quantum chemical calculations of aluminum substitution in zeolite ZSM-5[J].J.phys.chem.,1991,95(24):10031-10036.
[5]Liu Dan,Gui Jianzhou,Sun Zhaolin.Adsorp tion structures of heterocyclic nitrogen compounds over Cu(I)Y zeolite:a first principle study on mechanism of the denitrogenation and the effect of nitrogen compounds on adso rp tive desulfurization[J],J.mol.catal.A,2008,291(1):17-21.
[6]Liu Dan,Song Lijuan,Gui Jianzhou,et al.Adso rp tion structuresof heterocyclic sulfur compoundson Cu(I)Y zeolite:a first principle study[J].Stud.surf.sci.catal.,2007,170:1699-1704.
[7]Liu Dan,Gui Jianzhou,Liu Shi,et al.A density functional study of the chemisorption of thiophene on Cu(I)Y zeolite[J].Am.chem.soc.div.fuel chem.prep r.,2006,51(1):234-235.
[8]刘丹,张晓彤,桂建舟,等.Mo/HZSM-5催化剂表面活性中心的计算机模拟[J].兰州大学学报,2004,40(6):59-63.
Density Functional Theory Study of Acidity of RTH Structure
PENG Xi-lai,GU IJian-zhou,L IU Dan*,JIANG Hai-ying,YANG Shuang,GAO Yu-huan
(School of Chem istry and M aterials Science,L iaoning Shihua University,Fushun L iaoning113001,P.R.China)
The acidity of zeolite with RTH structure had been studied using the density functional theory generalized gradient approximation(GGA)method for the first time.The A l substitution energy of four different T sites and the dep rotonation energies of sixteen different Brönsted acid siteswere calculated to found the p referred A l substitution energy and acidic strength of different B acid sites.The results show that T4,locating in the opening(0.56 nm×0.25 nm)of 8-membered ring,is the most favo rable T site for A l substitution in the RTH structure.With considerations on both structure and acidity,A l4-O9(H)-Si2and A l4-O16(H)-Si4,locating in the intersection part of the two 8-membered ring channel,show stronger Brönsted acidity and are themost possible active sites for acid catalytic reaction.
RTH;Acidity;Zeolite;Density functional theory
O647
A
10.3696/j.issn.1006-396X.2011.02.003
2010-11-22
彭西来(1986-),男,江西吉安市,在读硕士。
国家自然科学基金项目(20706027);教育部重点项目(209031);辽宁省高等学校优秀人才支持计划项目(LR201024);辽宁省自然科学基金项目(20102131);辽宁省教育厅项目(2009A 430,L 2010243)。
*通讯联系人。
1006-396X(2011)02-0010-04
Received22N ovember2010;revised17December2010;accepted4M arch2011
*Corresponding author.Tel.:+86-13194131906;e-mail:ldan2000@163.com
