钙钛矿电子结构的密度泛函理论研究
2011-10-12孙兆林陈永昌王凌涛宋丽娟
徐 明, 孙兆林,2, 陈永昌,2, 王凌涛, 宋丽娟*
(1.辽宁石油化工大学辽宁省石油化工重点实验室,辽宁抚顺113001;2.兰州大学化学化工学院,甘肃兰州730000)
钙钛矿电子结构的密度泛函理论研究
徐 明1, 孙兆林1,2, 陈永昌1,2, 王凌涛1, 宋丽娟1*
(1.辽宁石油化工大学辽宁省石油化工重点实验室,辽宁抚顺113001;2.兰州大学化学化工学院,甘肃兰州730000)
应用密度泛函理论(DFT)计算方法,优化了K2Ti2O5的稳定几何构型,并计算了此钙钛矿体系的能带结构和态密度等基态物理性质。结果表明,K2Ti2O5属于间接绝缘体氧化物,其理论带隙宽度为2.6 eV。Ti原子处于氧原子的中心,其d轨道分裂为能量较高的eg和能量较低的t2g轨道,各轨道都靠近费米能级变为占据或半占据状态,而这对Ti原子的催化性能起着重要作用。分析了K2Ti2O5各个轨道电子转移的情况,为合成K2Ti2O5催化剂提供了一定的理论指导。
密度泛函理论; 钙钛矿; 催化; 态密度
众所周知,汽车尾气中的主要污染物为气体氮氧化物(NOx)和固体颗粒物(PM),它们的生成条件相互对立,导致汽车尾气排放本身存在NOx-PM之间你升我降的平衡关系。为了满足日益严格的排放法规,近10多年来国际上开始探索利用催化方法同时去除汽车尾气中的NOx和PM。
1972年,Voo rhoeve R J H等[1]报道了钙钛矿型氧化物在处理汽车尾气方面有广阔的应用前景。因此,具体特殊晶体结构的钛酸钾成为当前的研究热点,它被广泛的应用在高活性的催化、吸附等一系列的领域[2-7]。由于钛酸钾催化剂对同时消除PM和NOx具有很好的活性。因而人们将研究重点放在类钙钛矿和尖晶石型固定结构复合氧化物为主的催化剂上。钙钛矿催化剂的催化性能主要由B位离子决定,同时也间接受到A位离子的影响。A位取代会起到稳定晶体结构的作用,同时也可以改变B位阳离子的氧化态,从而改变氧空位数和阳离子缺陷,最终间接影响ABO3催化剂的性能。通过用较小的金属离子取代较大的A位金属离子,由于离子半径与电负性差异,使得氧离子在晶体中的迁移通道相对增大,可以有效地降低氧在晶格中的扩散位阻,提高氧的活动能力,从而促进氧与碳黑的接触,进而促进整个催化去除反应的进行。目前报道钛酸钾在固体颗粒物的氧化和氮氧化物的还原方面有举足轻重的作用。尽管在对于K2Ti2O5的实验研究已有较多的报道,但基于第一性原理的模拟计算分析则鲜有报道。
本文采用平面波超软赝势的方法对K2Ti2O5的电子结构进行了第一性原理的计算,从量子水平上研究了钛酸钾的能带结构和态密度等电子结构特性,探讨电子性质对K2Ti2O5选择性还原NOx和氧化PM的影响,为钛酸钾的改性提供了完善的理论基础。
1 理论模型和计算方法
1.1 理论模型
K2Ti2O5为单斜层状晶胞结构,如图1所示。其空间群为C2/m,晶胞参数a=1.137 nm,总共包含18个原子,其中4个Ti原子,4个K原子以及12个氧原子。材料结构优化后的晶格常数为a=1.083 nm,这是GGA计算固有的误差,不影响本文的分析计算。
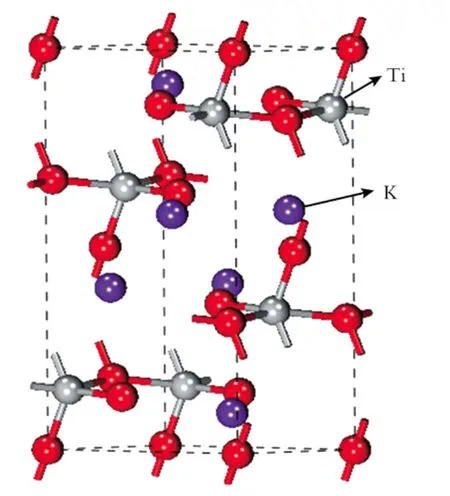
Fig.1 Calculated band structure of the K2Ti2O5图1 K2Ti2O5晶胞
1.2 计算方法
本文计算基于密度泛函理论(DFT)的CASTEP软件包。K,Ti和O三种原子价电子构型分别为3s23p64s1,3s23p63d24s2和2s22p4,各原子其它壳层的电子则当做芯电子处理。量子化学优化计算过程的具体计算参数设置如下:基函数选定为PW 91函数,对电子的交换相关能计算采用广义梯度进行近似(GGA)方法。截断波能量为350 eV,最大计算迭代次数为1 000,位移允许误差为0.005,布里渊区积分采用Monkhorst—Pack的特殊k点方案,选择2×2×3个网格点,在此条件下进行赝势和电荷密度的自洽迭代循环。迭代过程中的收敛精度为2×10-6eV/个,作用在每个原子上的力不大于0.01 eV/nm,内应力不大于0.1 GPa。
2 结果与讨论
图2为K2Ti2O5的能带结构。价带顶的位置在图2中仍对应能量值为零的点,这是因为软件包在计算宽禁带半导体或绝缘体时,把价带顶定义在费米能级(0 K),其他电子能级位置都是相对费米能级而言,这样处理的好处是便于对能带进行分析。由图2可以看出,K2Ti2O5的禁带宽度为2.6 eV,其导带底与价带顶位于Brillouin区的不同点,故属于间接带隙绝缘体。
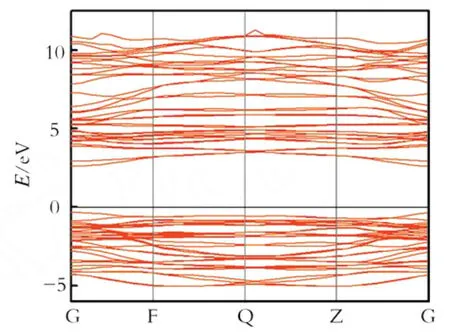
Fig.2 Calculated band structure of the K2Ti2O5图2 K2Ti2O5的能带结构
由电子态密度图可以分析电子在费米能级附近的行为,进而可得出各个原子轨道的相互作用及原子间的成键情况。图3和4分别为K2Ti2O5的态密度图(DOS)和各原子轨道的分态密度图(PDOS)。由图3和4可知,价带位于费米能级以下-5 eV<E<-0.2 eV区域,其主要源自氧原子的2p轨道,导带位于费米能级以上2.5 eV以上的范围,主要来自Ti原子的3d轨道。如图4所示,Ti原子的3d轨道被分为两大部分,根据晶体场理论,Ti原子的3d轨道被分裂为eg(dz2,dx2-y2)和t2g(dxy,dxz,dyz)电子轨道,相应的导带分裂为高导带和低导带,前者由Ti原子的eg电子轨道和O原子的2p电子轨道杂化而成,后者由Ti原子的t2g轨道与O原子的2p电子轨道杂化而成。相应的导带从图3可以看出,Ti原子和O原子在价带区明显的重叠,这表明Ti原子的3d轨道和O原子的2p轨道强烈的杂化,表现出共价键特性。然而最重要的是位于费米能级以下的轨道的杂化,因为这些杂化对化学键的成键起着至关重要的作用。同时也不难发现,态密度图波峰比较的尖锐,波峰跨度小,由此说明,钛酸钾原子之间的离域性较强,成键更强。
图5和6分别为Ti和O原子附近的电荷密度分布和差分电荷密度分布,其中O1,O2与O3分别代表不同位置的氧原子。由图5可以更直观的观察电荷的转移。图5表明钾原子在体态中是完全离子化的,Ti与O1,O2之间的有较强的结合作用,Ti-O以共价键的形式相互作用,这与前面的分波态密度图所描述的一致。差分电荷密度分布(Δρ)则直观地描述了表面区域电子重分布后各原子之间的成键状态,定义为空间某一点处,驰豫后该体系所有原子的电荷密度分布函数的总和(∑ρA)与驰豫前的体系的电荷密度(ρS)的差分。其表达式见式(1)。
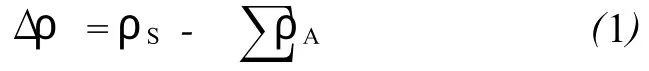
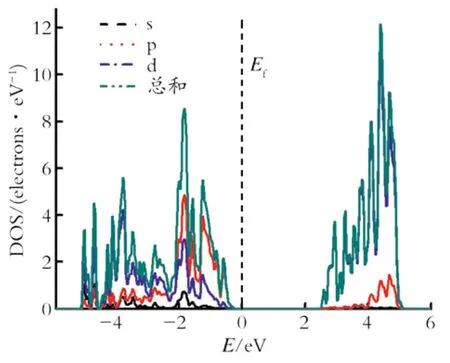
Fig.3 Projected density of states of K2Ti2O5图3 K2Ti2O5的态密度
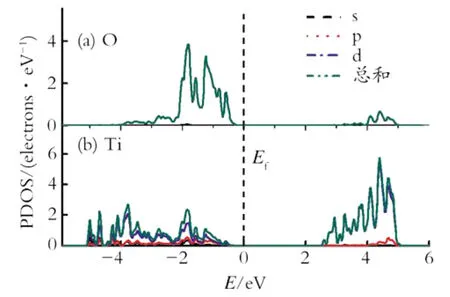
Fig.4 O,Tiatom partial density of states图4 O,Ti原子的分波态密度
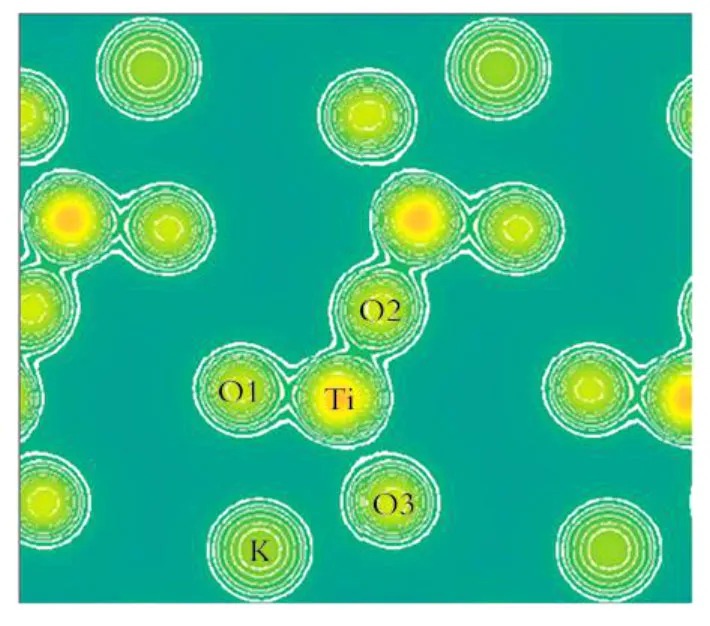
Fig.5 Charge density of K2Ti2O5图5 K2Ti2O5的电荷密度
图6可以观察到Ti原子周围出现了较强的亮点,即这个区域电荷密度变化较大,可以说明Ti原子与氧原子之间发生了电子转移,因此Ti原子沿d轨道失去电荷的同时又从其他的轨道得到电荷,这与前面的分波态密度图所描述的一致,正是由于在不同成键方向上共价键强弱的差异,能够使钛酸钾表现出很大的活性,并且通过吸附能得到比较稳定的结构。同时还可以观察到与Ti原子相连接的钾原子几乎没有发生电子的转移。结合以上方面,我们可以得出Ti-O之间的相互作用,Ti的2t2g是由t2g与O2p的轨道的重叠而成,3eg为他们的相互作用而成,其电荷的转移对共价键的混合程度起着重要的作用。因为,Ti的3d轨道与O2p具有相对稳定的共价性,可以有效地减少3d电子的定域性,电子更容易从轨道中逃逸而跃迁,从而使催化活性也随之升高[8]。
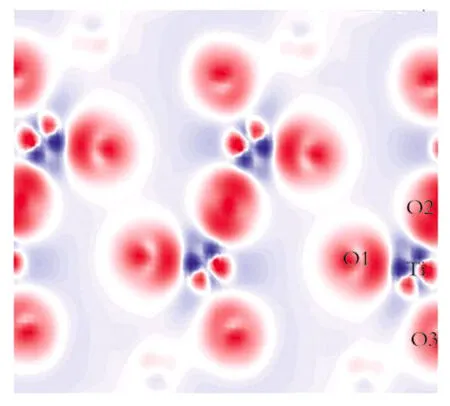
Fig.6 Charge density difference of K2Ti2O5图6 K2Ti2O5的差分电荷密度
3 结束语
基于密度泛函理论计算了催化剂K2Ti2O5的的电子结构,并通过对其能带结构、态密度、电荷密度和差分电荷密度的分析,结果表明,K2Ti2O5属于间接绝缘体氧化物,其理论带隙宽度为2.6 eV。其价带主要是由Ti的3d轨道所构成,氧的2p轨道也有贡献。Ti原子处于氧原子的的中心,其d轨道分裂为能量较高的eg和能量较低的t2g轨道,各轨道都靠近费米能级变为占据或半占据状态,而这对Ti原子的催化性能起着重要作用,通常费米能级以下区域的杂化对体系起着非常重要的作用。通过对钛酸钾电子结构第一性原理的计算,为其催化反应机理的研究提供了理论的指导。
[1]Voorhoeve R J H,Remeika J P,Freeland P E,et al.Rare-earch oxides of managanese and cobalt rival platinum for the treatment of carbon monoxide in auto exhaust[J].Science,1972,177:353-354.
[2]Yahya R B,Hayashi H.Hydrothermal synthesis of potassium hexatitanates under subcritical and supercritical water conditions and its application in photocatalysis[J].Chem.mater.,2001,13(3):842-847.
[3]Bao N,Feng X,Yang Z,et al.Highly efficient liquid-phase photooxidation of an azo dye methyl o range over novel nanostructured po rous titanate-based fiber of self-supported radially aligned H2Ti8O17·1.5H2O nanorods[J].Sci.technol.,2004,38(9):2729-2736.
[4]Yin S,Sato T.Synthesis and photocatalytic properties of fibrous titania prepared from protonic layered tetratitanate precursor in supercritical alcohols[J].Ind.eng.chem.,2000,39(12):4526-4530.
[5]Corcoran D JD,Tunstall D P,Irvine J T S.Hydrogen titanates as potential proton conducting fuel cell electrolytes[J].Solid state ionics,2000,136:297-303.
[6]Sasaki T,Izumi F,Watanabe M.Intercalation of pyridine in layered titanates[J].Chem.mater.,1996,8(3):777-782.
[7]Sikuvhihulu L C,Coville N J,N tho T,et al.Potassium titanate:An alternative support for gold catalyzed carbon monoxide oxidation[J].Catal.lett.,2008,123:193-197.
[8]A rima T,Tokura Y,Tottance JB.Variation of optical gaps in perovskite-type 3d transition-metal oxides[J].Physical rewiew B,1993,48(23):17006-17009.
(Ed.:SGL,Z)
Density Functional theory Study of Perovskite with Electronic Structure
XU M ing1,SUN Zhao-lin1,2,CHEN Yong-chang1,2,WANG Ling-tao1,SONG Li-juan1*
(1.L iaoning Province Key Laboratory of Petrochem ical,L iaoning Shihua University,Fushun L iaoning113001,P.R.China;2.Chem istry and Chem ical Engineering,Lanzhou University,Lanzhou Gansu730000,P.R.China)
Density functional theory(DFT)calculations were carried out for the stability op timization of K2Ti2O5.Band structure and density states with other ground states physical properties of the perovskite system were calculated.The results show that K2Ti2O5is indirect insulator oxides and the theoretical band gap width is 2.6 eV.Ti atom is in the center of the oxygen atom,the dorbital splits for the higher energy egorbit and the lower energy t2gorbit,the orbits are close to the Fermi level becomes occupied or semi-occupied state,which p lays an important role for Ti atom s with the catalytic Performance.Analysed each orbit of K2Ti2O5with electron transfer order to the synthesis of K2Ti2O5provides some theoretical guidance.
Density functional theory;Perovskite;Catalysis;State density
O643.36
A
10.3696/j.issn.1006-396X.2011.02.010
2011-02-21
徐明(1984-),男,山东潍坊市,在读硕士。
辽宁省教育厅资助项目(2009T061)。
*通讯联系人。
1006-396X(2011)02-0040-03
Received21February2011;revised12A pril2011;accepted15A pril2011
*Corresponding author.Tel.:+86-13941350056;e-mail:lsong@lnpu.edu.cn
