利巴韦林注射液半成品质量控制方法优化
2011-09-20巩新玉杨利民
巩新玉 闫 晗 刘 静 赵 璨 杨利民
(1 驻马店市食品药品检验所,河南 驻马店 463000;2 周口市太康人民医院,河南 周口 463000)
利巴韦林的化学名称:1-β-D-呋喃核酸基-1H-1,2,4-三氮唑-3-羧酰胺。
利巴韦林的结构式[1]:
分子式:C8H12N4O5
分子量:244.21

利巴韦林(Ribavirin)又称三氮唑核苷、病毒唑、三唑核苷。为广谱抗病毒核苷类化合物,能抑制病毒合成核酸,对多种RNA、DNA病毒有抑制作用。临床用于病毒性感冒、腺病毒、肺炎麻疹、甲型肝炎、流行性出血热、带状疱疹及病毒性脑炎等疾病的治疗[2,3]。
HPLC法为中国药典2005年版二部规定的利巴韦林(原料及注射液)含量测定的方法,专属性强,准确可靠,重复性好,适用于利巴韦林注射液成品质量控制。因利巴韦林注射液在生产过程中,从配制到灌封不能超过12h,HPLC法消耗时间过长,流动相的配制复杂,成本较高,需要对样品进行过滤和超声等前处理。利巴韦林注射液半成品的质量检测项目有pH值和含量测定,pH值的测定方法在此不做讨论,主要讨论含量测定。利巴韦林的含量测定的方法有定氮法 、液相色谱法[1]、反向高效液相色谱法、高效毛细管电泳法、旋光法[4]、紫外分光光度法[3]、高磺酸法等。这里对几种常见简便的方法进行比较,优化出一种适合生产中利巴韦林注射液半成品质量控制的方法。
从利巴韦林分子结构上可知具有紫外吸收性和旋光性,这与中国药典2005年版二部上的利巴韦林性质一致。
利巴韦林注射液处方:利巴韦林100g;氯化钠9g;药用炭0.5g;注射用水加至1000mL。
在旋光光度测定方法中,通过对辅料进行旋光检测,辅料没有旋光度,对此种方法进行含量测定没有影响。再分别用以上两种方法测定利巴韦林注射液半成品的含量并与HPLC法测得含量进行对比优化出一种能够保证准确性,节省时间,成本低,适合于利韦林注射液生产需要的方法对利巴韦林注射液半成品进行控制,并进行方法学考察。
1 实验仪器与试剂
SSI2000型HPLC-UV:(美国联盟实验室);AL104/01型电子天平:(瑞士梅特勒);pHS 3C精密pH计(上海雷磁仪器厂);UV-1201紫外可见分光光度计(北京通用仪器设备公司);WZZ-1型自动指示旋光仪(上海物理光学仪器厂);利巴韦林对照品(中国药品生物制品检定所);利巴韦林原料:(济南明鑫制药有限公司)【批号】 ;氯化钠:(中盐宏博集团云梦云虹制药有限公司)【批号】;药用炭:(上海活性炭有限公司);利巴韦林注射液半成品规格:0.1mg/mL(濮阳市汇元药业有限公司)【批号:081101,081102,081103】。
实验用水均为超纯水
2 实验方法与结果
2.1 旋光光度法
2.1.1 检测条件
利巴韦林具有旋光性,为左旋性物质,采用钠光谱D线,波长为589nm 测定管长为20cm在20℃±0.15℃,以水为溶剂。
2.1.2 旋光度与浓度的线性关系
精密称取经105℃干燥至恒重的注射用利巴韦林,加注射用水,分别配成40、80、100、120、140mg/mL的5种浓度的溶液,依照2.1.1中的方法测定旋光度。以旋光度为纵坐标,浓度为横坐标进行线性回归,得到方程Y =-0.0743X-0.0981,(r=0.9999)。数据和线性图见表1和图1。

表1 标准曲线的制备
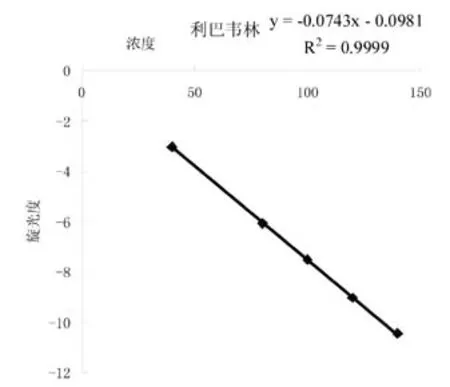
图1 标准曲线(旋光)
2.1.3 精密度实验
取以上5种浓度的利巴韦林溶液,照旋光光度测定法[1]测定旋光度,重复测定6次,计算标准相对偏差,见表2。
2.1.4 稳定性实验
2.1.4.1 温度对旋光度的影响
取不同温度的以上5种浓度的溶液,分别测定旋光度结果,见表3。实验结果表明,在10~30℃范围温度内对利巴韦林溶液的旋光度基本没有影响。
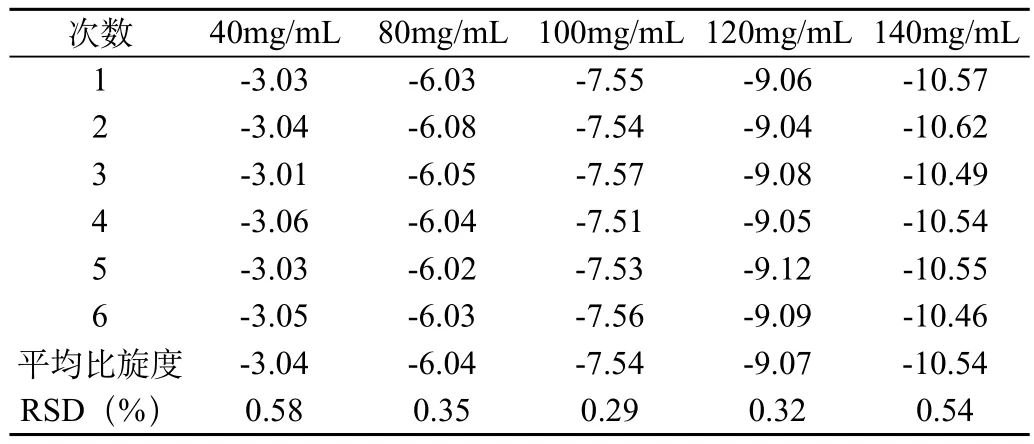
表2 精密度实验
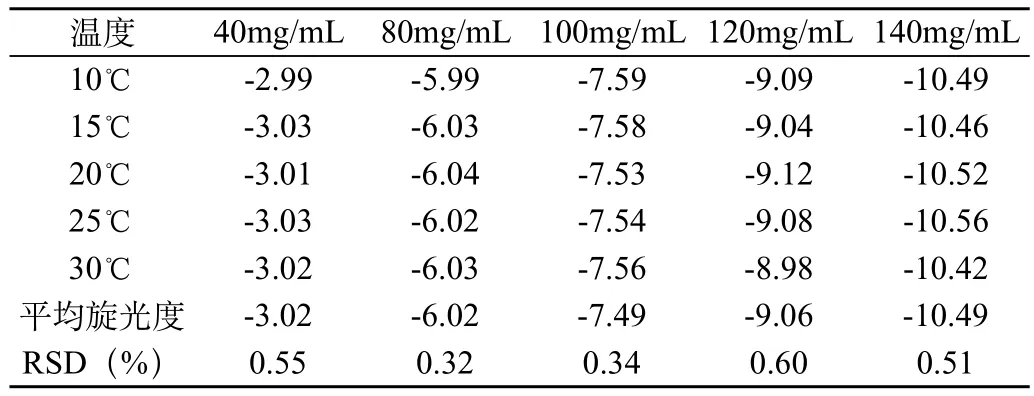
表3 温度对旋光度的影响
2.1.4.2 pH值对旋光度的影响
药典规定利巴韦林注射液pH值范围是4.0~6.0,取浓度为40mg/mL的利巴韦林溶液,分别用0.1mol/L的盐酸溶液和0.1mol/L的氢氧化钠溶液调整至不同的pH值(4.0、4.5、5.0、5.5、6.0),测定旋光度,见表4。实验结果说明pH值在4.0~6.0范围内,利巴韦林注射液旋光度无变化。

表4 pH值对旋光度的影响
2.1.5 回收率实验
根据以上配制方法配制浓度为40、80、100mg/mL的利巴韦林溶液,每种浓度的溶液各3份,根据中国药典2005年版二部的旋光光度法,测定旋光度,并计算含量,结果见表5。
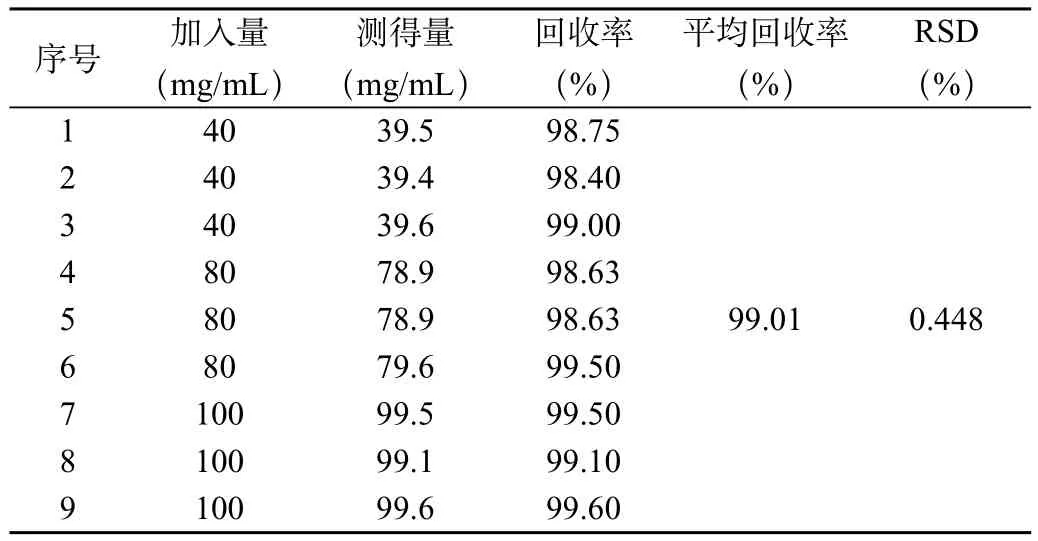
表5 回收率试验
2.1.6 样品的含量测定
取以上081101、081102、081103,三批利巴韦林注射液半成品,直接根据旋光光度法测定旋光度,并计算含量,见表6两种方法含量比较。
2.2 HPLC法
2.2.1 色谱条件
色谱柱为Hamilton HC-75 氢型阳离子交换柱(250×4.1mm,9μm);流动相为超纯水(pH用稀硫酸调至2.5±0.1);流速为1mL/min,紫外检测波长为207nm,柱温25℃,检测理论塔板数按利巴韦林峰计算不低于3000。
2.2.2 流动相的配制
取蒸馏水2000mL,加稀硫酸调节pH值到2.5,用0.45µm的滤膜抽滤,弃去初滤液,取续滤液,在超声波中处理20min,作为流动相。
2.2.3 对照液的制备
取利巴韦林标准品12.5mg,精密称定,置50mL的量瓶中,用流动相稀释至刻度,摇匀。精密量取稀释液5mL,置25mL的量瓶中,用流动相稀释至刻度,摇匀。
2.2.4 样品含量的测定
取以上081101、081102、081103三批利巴韦林注射液半成品,精密量半成品5mL置100mL量瓶中,用流动相稀释到刻度,摇匀。量取稀释液1mL,置100mL量瓶中,加流动相稀释到刻度,摇匀后,用0.45µm的滤膜抽滤,弃去初滤液。取续滤液,超声波处理后按照上述色谱条件,精密量取20µL注入液相色谱仪,记录峰面积。与标准品的峰面积对比,计算含量。见图2和图3。

图2 081103批半成品扫描图谱
图3 标准品扫描图谱
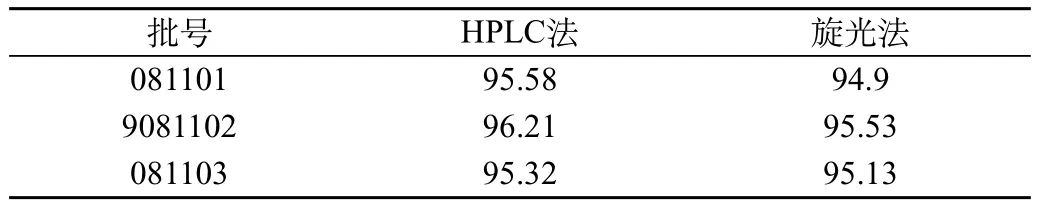
表6 两种含量测定方法的比较
把HPLC法和旋光法测得利巴韦林注射液半成品的含量同样进行t检验,经计算t=1.5719。自由度df=(N1+N2-2)=4,查表[11]得t0.05(4)=2.776。t<t0.05(4),没有显著性,说明旋光法可采用。
3 讨论与分析
利巴韦林注射液的辅料为氯化钠,在辅料对主药吸收度的影响中附有扫描光谱,可见辅料对含量测定结果不产生实质性影响。为了简化试验操作,可以用蒸馏水作为空白。
根据处方的比例,配制成不含利巴韦林的对照液,经过旋光测定,证明没有旋光性,辅料对主药含量测定没有影响。进行含量测定时可直接用蒸馏水作为对照校正。
通过对以上方法的含量测定结果分别与HPLC比较,旋光光度法的含量测定结果偏低。通过进行数理学T检验证明,没有显著性差异,两种方法都可采用。
4 结 论
通过对以上方法的整体考察,旋光光度法与HPLC相比。HPLC法专属性强,准确可靠,重复性好,但不适合利巴韦林注射液半成品的质量控制。旋光光度法比HPLC法相对节省了时间,操作更方便,均可用于利巴韦林注射液半成品的质量控制。
旋光光度法样品检测浓度不要求太低,利巴韦林注射液为0.1g/mL,浓度适宜,可直接进行旋光测定,此浓度测定结果误差较小。其操作简单,可直接用对照品测得校正因子后,照2005版中国药典《旋光度测定法操作规程》依法测定,按下式计算,含利巴韦林(C18H12N4O5)应为标示量的93.0~107.0%。
含量%=α×校正系数×100%
此方法简便,快速,适合本厂生产需要,缩短了配制到灌封的时间。旋光光度法是为适应本厂优化出来的较为理想的方法,通过此方法的优化,减少了化验人员的劳动强度,降低了成本。
[1]国家药典委员会.中华人民共和国药典2005年版(二部)[S].北京:化学工业出版社,2005:262.
[2]杜兴,何英梅,罗西湘.利巴韦林片含量测定方法改进[J].中国药事,2006,20(3):171-172.
[3]林婉贞,刘怡,任斌,等.紫外分光光度法测定利巴韦林含片含量[J].天津药学,2005,17(2):29-31.
[4]范宇,王联民.旋光法测定利巴韦林注射液含量方法探讨[J].徐州医学院学报,2005,25(2):152-154.
[5]李安荣,许群芬,龚亚林.紫外分光光度法和高效液相色谱法测定利巴韦林片含量的比较[J].中国药业,2004,13(12):34-35.
