3-苯并噁唑-6-甲酰基咔唑的合成
2011-07-30费学宁谷迎春
杨 旭,费学宁,谷迎春,李 超
(天津城市建设学院 材料科学与工程系,天津 300384)
咔唑及其衍生物具有较大的共轭体系及强的分子内电子转移性能[1],其作为重要的精细化学品中间体被广泛应用于染料、医药、农药、香料、高分子等领域[2-3].设计合成新型的咔唑衍生物成为近年来研究的热点之一.
苯并噁 唑类化合物有较强的吸电子能力,并具有强烈的荧光,有的还具有激光性能,可以用作光致发光材料、增白剂、激光染料和荧光探针[4-5].同时,苯并 噁唑基及其衍生物由于其特殊的生理活性而被广泛应用于医药、农药、生物化学等领域,如作为杀菌剂[6]、除草剂[7]、抗肿瘤剂[8]等.
苯并噁 唑基是含有N,O杂原子的杂环化合物,具有优秀的推拉电子能力,将其引入咔唑中增长了分子共轭链,保持了分子刚性共轭平面结构,使得整个共轭单元分子的电荷重新分布,从而有利于提高分子的光学性能.为此笔者设计并合成了2个3-苯 并噁 唑-6-甲酰基咔唑衍生物(合成路线见图1),其结构经1H NMR和MS表征,并初步研究了其荧光光谱.

图1 目标化合物合成路线
1 实验部分
1.1 仪器与试剂
所用试剂均为市售分析纯.仪器:GF254型薄层色谱硅胶(青岛海洋化工有限公司);柱层析色谱硅胶(青岛源博硅胶有限责任公司);X-4数字显微熔点测定仪(温度计未经校正,北京泰克仪器有限公司);Bruker AC-P400(400 MHz)型核磁共振仪,TMS为内标;质谱仪ESI 离子源(美国热电公司);紫外-可见分光光度计(UV2550,日本岛津);荧光分析仪 Cary Eclitse(美国 VARINA).
1.2 合成方法
1.2.1 N-取代咔唑(1)的合成[9]
化合物1a的合成步骤参考文献[9].
N-苄基咔唑 1 b的合成步骤同1a,白色针状晶体,产率 71.3%,m.p.113~115 ℃.1H NMR(400 MHz,CDCl3)δ:5.50(2H,s),7.12(2H,d,J=6.80 Hz),7.22~7.26(5H,m),7.35(2H,d,J=8.40 Hz),7.40~7.44(2H,t,J=7.40 Hz),8.13(2H,d,J=7.60 Hz).MS(ESI)m/z:258(M++1).
1.2.2 3-甲酰基-N-取代咔唑(2)的合成[9]
化合物2a的合成步骤参考文献[9].
3-甲酰基-N-苄基咔唑2b的合成步骤同2a,白色固体,产率 68.6%,m.p.127~129 ℃.1H NMR (400 MHz,CDCl3)δ:5.52(2H,s),7.12(2H,d,J=5.60 Hz),7.25(3H,s),7.31~7.35(1H,t,J=7.20 Hz),7.39~7.43(2H,t,J=8.00 Hz),7.47~7.50(1H,t,J=7.40 Hz),7.95(1H,d,J=8.40 Hz),8.17(1H,d,J=7.60 Hz),8.62(1H,s),10.07(1H,s,CHO).MS(ESI) m/z:286(M++1).
1.2.3 咔唑席夫碱(3a)的合成
将 1.2 g(5.2 mmol)3-甲酰基-N-乙基咔唑和0.8 g(8.0 mmol)邻氨基苯酚溶于40 mL无水乙醇中,回流,滴加5滴冰乙酸,反应4 h.反应完毕冷却至室温,将反应液缓慢倒入300 mL冰水中,析出黄色固体,过滤,冷乙醇洗涤,得黄色黏稠固体0.8 g,产物不经提纯直接进行下一步.
化合物3 b的合成同上,黄色固体,产物不经提纯直接进行下一步.
1.2.4 3-苯并噁唑基-N-乙基咔唑(4a)的合成
将1.2 g(3.9 mmol)席夫碱3a和2.1 g(9.6 mmol)三价醋酸锰溶于 50 mL的 DMSO 中,140 ℃反应24 h.反应完毕冷却至室温,将反应液缓慢倒入500 mL冰水中,析出红棕色固体,抽滤,洗涤,柱层析分离,得淡黄色固体 0.6 g,产率 52.4%,m.p.141~143 ℃.1H NMR(400 MHz,CDCl3)δ:1.43~1.48(3H,t,J=7.20 Hz),4.33~4.39(2H,m),7.30~7.33(3H,m),7.43(1H,d,J=8.70 Hz),7.46~7.50(2H,m),7.56~7.59(2H,m),8.18(1H,d,J=7.80 Hz),8.32~8.35(1H,m),8.97(1H,s).MS(ESI)m/z:313(M++1).
3- 噁苯并 唑基-N-苄基咔唑 4b的合成步骤同4a,白色固体,产率 60.4%,m.p.170~172 ℃.1H NMR(400 MHz,CDCl3)δ∶5.60(2H,s),7.19(2H,d,J=6.80 Hz),7.29~7.32(3H,t,J=6.40 Hz),7.35~7.38(3H,t,J=6.80 Hz),7.44(1H,d,J=8.00 Hz),7.51(2H,d,J=8.40 Hz),7.64(1H,d,J=8.00 Hz),7.82(1H,d,J=8.40 Hz),8.26(1H,d,J=7.60 Hz),8.37(1H,d,J=8.40 Hz),9.09(1H,s).MS(ESI)m/z:375(M++1).
1.2.5 3-苯并噁唑基-6-甲酰基-N-乙基咔唑(5a)的合成
在100 mL单口烧瓶中加入 DMF(7.7 mL,100 mmol),冰水浴冷却下,缓慢滴加POCl3(9.5 mL,100 mmol),溶液呈微红色.室温下,向其中缓慢滴加3- 噁苯并 唑基-N-乙基咔唑(3 g,10 mmol)的1,2-二氯乙烷溶液40 mL,滴毕,回流反应10 h.冷却至室温,将反应液缓慢倒入500 mL冰水中,二氯甲烷萃取,有机相水洗,干燥.柱层析分离得淡黄色固体 0.4 g,产率 13.3%,m.p.257~260 ℃.1H NMR(400 MHz,CDCl3)δ:1.48~1.52(3H,t,J=7.60 Hz),4.37~4.43(2H,m),7.37~7.40(2H,m),7.47~7.52(2H,m),7.60(1H,d,J=9.60 Hz),7.79(1H,d,J=9.60 Hz),8.04(1H,d,J=9.60 Hz),8.38~8.40(1H,d,J=10.80 Hz),8.63(1H,s),8.79(1H,s),10.10(1H,s,CHO).MS(ESI)m/z:341(M++1).
3- 噁苯并 唑基-6-甲酰基-N-苄基咔唑5 b的合成步骤同 5a,白色固体,产率 20.7%,m.p.212~214℃.1H NMR(400 MHz,CDCl3)δ:5.58(2H,s),7.15(2H,d,J=6.40 Hz),7.30(3H,d,J=6.40 Hz),7.35~7.37(2H,t,J=7.20 Hz),7.46~7.52(2H,m),7.60(1H,d,J=7.60 Hz),7.78(1H,d,J=4.80 Hz),8.02(1H,d,J=8.40 Hz),8.38(1H,d,J=8.00 Hz),8.70(1H,s),9.05(1H,s),10.11(1H,s,CHO).MS(ESI)m/z:403(M++1).
2 结果与讨论
2.1 合成
咔唑N上有一对孤对电子,使得溴乙烷和溴化苄中溴离去后的碳很容易与咔唑上的 N发生亲核取代反应.甲酰化反应中使用DMF作为甲酰化试剂,1,2-二氯乙烷作为溶剂,反应时间10 h左右为最佳条件.席夫碱氧化所用的三价醋酸锰为自制,冰乙酸在使用前需经高锰酸钾精制以除去其中的还原性物质.
2.2 核磁共振
化合物 4的核磁共振波谱中,化学位移为8.97×10-6的单峰归属于咔唑环 4′位上的氢,在化合物 5 的核磁共振波谱上有化学位移为 8.79×10-6的单峰与之相对应,另外化合物 4 的化学位移中(7.46~7.59)×10-6的多重峰由 4个氢变为化合物5的(7.47~7.60)×10-6的 3个氢,其中 1个氢被甲酰基取代,而10.10×10-6的新峰为甲酰基上的活泼氢的化学位移,证明了化合物 5的结构是正确的,也间接地为化合物3的结构表征提供了重要佐证.化合物5 a和5 b的1H NMR类似,其他相应官能团的位移信号在谱图中都能得以确认.
2.3 荧光光谱
将化合物5a和5b分别溶于二氯甲烷,配置成相同浓度的溶液,在激发波长为360 nm的条件下,测定化合物5a和5b的荧光光谱,如图2所示.化合物5a和5b的发射波长分别为403.9,414.1 nm,后者相对于前者红移了10.2 nm,荧光强度增强了157.这主要是由于苯并噁 唑的引入扩展了分子的共轭链,使得分子中具有较大的共轭π键结构.N位上含有苄基的化合物5b与N位上含有乙基的化合物5a相比,发射波长发生红移,红移不多,发光强度增强,说明苄基的引入有可能使整个分子的共轭体系增大,对分子的荧光发射波长和对荧光强度稍有影响.
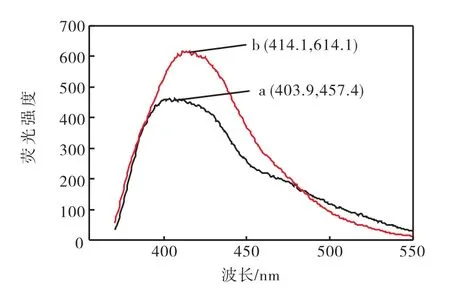
图2 化合物5a和5b的荧光发射光谱
3 结 论
本文设计合成了2个3- 噁苯并 唑-6-甲酰基咔唑衍生物,其结构经过1H NMR,MS表征,并将2个咔唑衍生物荧光光谱进行了对比.结果表明,相同浓度下,化合物 5 a最大吸收波长为 360 nm,最大发射波长为 403 nm,Stocks位移为 43 nm,相对于化合物5a,化合物 5b的最大发射波长增大了 10 nm,Stocks位移增大了11 nm,荧光强度也有所增加.两个目标化合物的合成为研究光电材料和光致变色材料提供了基础,可以作为发光材料化合物合成的中间体,此外,还可以作为制备荧光探针的中间体,进而合成应用于生物体系的荧光探针分子.
[1] 陈光需,吴恒富,骆俊山,等. 一种D-π-A型咔唑类衍生物的合成[J]. 合成化学,2008,16(4):431-433.
[2] 赵德丰,周丹红,杨锦宗. 咔唑化学的进展及其在颜料工业中的应用[J]. 染料与染色,1987(4):32-38.
[3] Gingrich DE,Reddy DR,LQBAL MA. et al. A new class of potent vascular endothelial growth factor receptor tyrosine kinase inhibitors:structure-activity relationships for a series of 9-alkoxymethyl-12-(3-hydroxypropyl)indeno[2,1-a] pyrrolo[3,4-c] carhazole 5-ones and the identification of CEP-5214 and its dimethylglycine ester prodrug clinical candidate CEP-7055[J]. Journal of Medicinal Chemistry,2003,46(25):5 375-5 388.
[4] KATARZYNA G,MARIUSZ S,JOANNA M,et al.Synthesis and photophysical properties of 3-[2-(pyridy1)benzoxazol-5-y1]-L-alanine derivateives [J].Tetrahedron,2002,58(11):2 201-2 209.
[5] IRINA P,PETER N,VENETA D. Photophysical properties and quantum chemical calculations of differently substituted-(2-phenyletheny1)-benzoxazoles and benzothiazoles[J]. Journal of Photochemistry and Photobiology A:Chemistry,2000,133(1/2):21-25.
[6] RAJENDRA S,VARMA,CHAUHAN S. Synthesis of substitued 2-Phenylbenzoxazoles as CNS agents [J]. Indian Journal of Chemistry,1995,24B(3):280-285.
[7] 宜 兵,林原斌. 噁双苯并 唑二苯乙烯的合成[J]. 化学试剂,2001,23(3):176-177.
[8] 王振宁,胡德禹,宋宝安,等. 噁具有杀菌活性的 唑类衍生物的研究进展[J]. 精细化工中间体,2008,2(4):9-15.
[9] 李 超,费学宁,谷迎春,等. 3-苯并噻唑-6-甲酰基咔唑的合成[J]. 合成化学,2010(S1):40-43.
