反相高效液相色谱法测定盐酸米诺环素原位凝胶中盐酸米诺环素的含量
2011-07-25杨跃辉郜琪臻丁平田
杨跃辉,郜琪臻,丁平田
(1.中国医科大学附属盛京医院药学部,辽宁 沈阳 110004;2.中国医科大学附属第一医院药学部,辽宁 沈阳 110001;3.沈阳药科大学药学院,辽宁 沈阳 110015)
盐酸米诺环素(Minocycline hydrochloride,MINO·HCl),又名美满霉素、盐酸二甲胺四环素、米诺四环素,为四环素族抗生素。盐酸米诺环素是广谱的抑菌剂,能抑制大部分牙周病原菌[1];体外实验证明,盐酸米诺环素的抑菌效果较其它抗菌药好,临床定期应用盐酸米诺环素可明显减少微生物的数量[2]。将盐酸米诺环素制成微胶囊型牙周缓释原位凝胶,一周给药一次,具有明显的缓释效果和确切的疗效,可使药物在牙周袋内局部释放,减少给药剂量,提高局部药物浓度,延长药物在牙周袋内的作用时间。
作者在此采用反相高效液相色谱(RP-HPLC)法测定盐酸米诺环素原位凝胶中盐酸米诺环素的含量。
1 实验
1.1 试剂与仪器
盐酸米诺环素(MINO·HCl)、甘油(GLY)、羟乙基纤维素(HEC)、甘油三醋酸酯(TA)、氨烷基甲基丙烯酸酯共聚物、乙酸铵、氯化钾(广东汕头西陇化工厂)、乙二胺四乙酸二钠,分析纯;乙腈、甲醇,色谱纯;去离子水。
Sartorius BS 110S型电子分析天平,北京赛多利斯仪器系统有限公司;立式扩散仪,上海锴凯科技贸易有限公司;ZD-85A型恒温气浴振荡器、85-2A 型恒温磁力加热搅拌器,江苏省金坛市荣华仪器制造有限公司;高效液相色谱仪、2201 型紫外扫描分光光度计,日本岛津公司;半透膜,美国Biosharp公司;KQ-50B型超声波清洗器,昆山市超声仪器有限公司。
1.2 方法
1.2.1 色谱条件
色谱柱:Kromasil C8(4.6 mm×250 mm,5 μm);流动相为乙酸铵缓冲盐溶液-乙腈(250∶105);检测波长350 nm;流速1.0 mL · min-1;柱温35 ℃;进样量10 μL。
1.2.2 系统适应性考察
取盐酸米诺环素约10 mg,置于25 mL量瓶中,加水5 mL使溶解后,置沸水浴中加热60 min,冷却,加水稀释至刻度,摇匀,取10 μL注入高效液相色谱仪,记录色谱图。
1.2.3 标准曲线的绘制
精密称取盐酸米诺环素适量,置于100 mL量瓶中,加甲醇40 mL,振摇使溶解,加流动相定容至刻度,即得盐酸米诺环素标准贮备液。
分别取盐酸米诺环素标准贮备液适量,配制成不同浓度的盐酸米诺环素标准溶液(分别相当于米诺环素50 μg · mL-1、100 μg · mL-1、300 μg · mL-1、500 μg · mL-1、750 μg · mL-1、1000 μg · mL-1)。精密量取盐酸米诺环素标准溶液10 μL注入高效液相色谱仪,记录色谱图。绘制标准曲线,如图1所示。拟合线性回归方程为:A=5327c+ 19583(R=0.9999)。结果表明,盐酸米诺环素在50~1000 μg · mL-1(以米诺环素计)范围内与峰面积呈良好的线性关系。
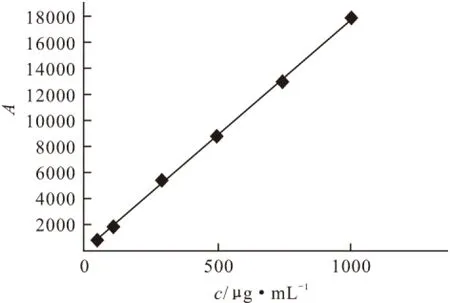
图1 盐酸米诺环素的标准曲线
1.2.4 精密度实验
按1.2.3方法分别配制高、中、低三个浓度(相当于米诺环素1000 μg·mL-1、500 μg·mL-1、50 μg·mL-1)的盐酸米诺环素溶液各5份,置于4℃冰箱中,精密量取10 μL注入高效液相色谱仪中,记录液相色谱图。由标准曲线回归方程计算其浓度测定值,考察日内和日间精密度。
1.2.5 回收率实验
取空白原位凝胶适量,置于100 mL量瓶中,加甲醇40 mL,充分振摇,超声助溶30 min,直至未见有胶状物质存于量瓶底部。精密称取盐酸米诺环素适量(相当于米诺环素1000 μg · mL-1、500 μg · mL-1、50 μg · mL-1)。加流动相稀释至刻度,摇匀,过滤,取续滤液作为供试品溶液。精密量取10 μL供试品溶液注入高效液相色谱仪中,记录色谱图。由标准曲线回归方程计算其浓度测定值,计算回收率。
2 结果与讨论
2.1 检测波长的确定
精密称取盐酸米诺环素适量,用甲醇溶解,配制成10 μg · mL-1的溶液,以甲醇为参比,在200~400 nm波长范围内扫描。结果显示,盐酸米诺环素甲醇溶液在250 nm、350 nm处有最大吸收。
精密称取空白原位凝胶适量,加适量甲醇,充分振摇,超声助溶30 min,用微孔滤膜过滤,取续滤液,以甲醇为参比,在200~400 nm波长范围内扫描。结果显示,空白原位凝胶在350 nm处无吸收。故选择350 nm作为检测波长。
2.2 系统适应性考察(图2)

1.差向米诺环素 2.米诺环素
由图2可知,米诺环素峰的拖尾因子应在0.9~1.35之间,米诺环素峰与差向米诺环素峰(相对保留时间约为0.8 min)的分离度应不小于2.5[3]。
2.3 精密度实验(表1)
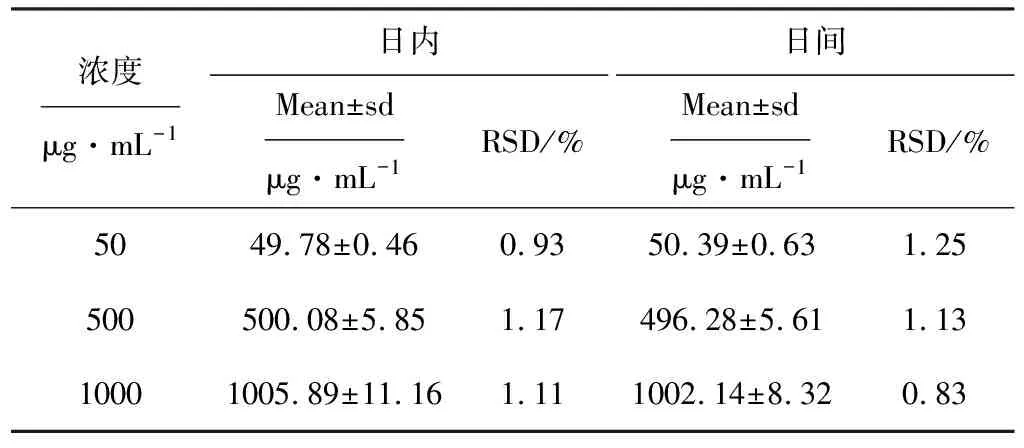
表1 精密度实验结果(n=5)
由表1可知,日内和日间精密度的相对标准偏差均小于2%,符合方法学要求。
2.4 回收率实验(表2)
由表2可知,回收率符合方法学要求。
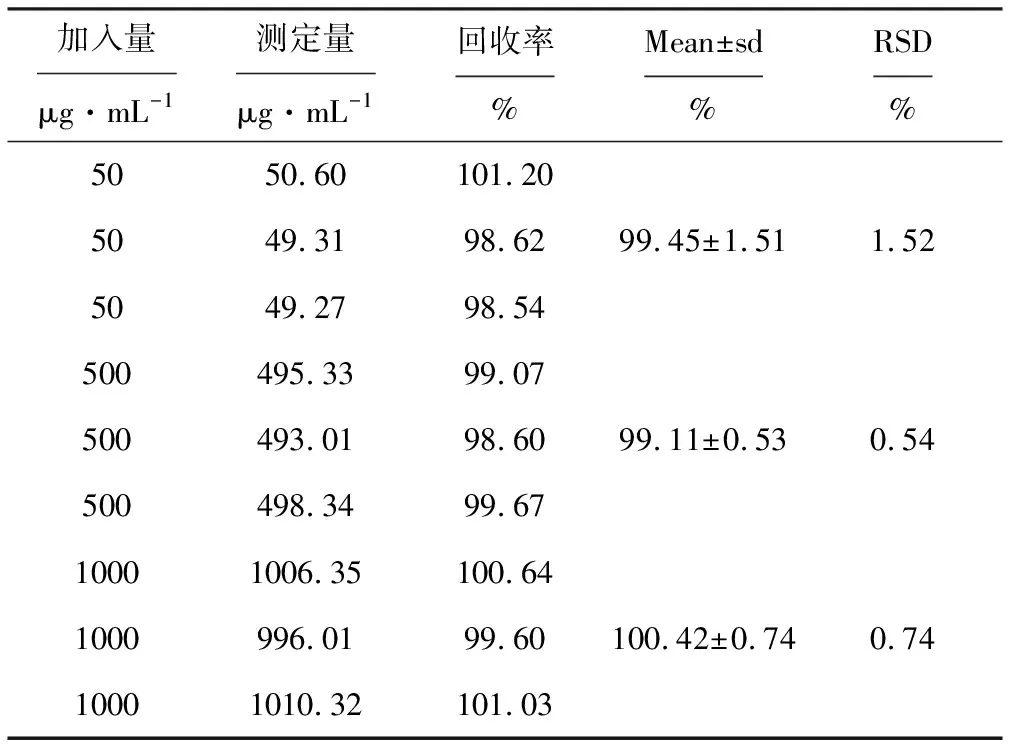
表2 回收率实验结果
2.5 最低检测限
配制一系列不同浓度的盐酸米诺环素溶液,精密量取10 μL注入高效液相色谱仪中,记录色谱图,得到最低检测限为10 ng · mL-1(S/N≥3)、定量限为50 ng · mL-1(S/N>10)。
2.6 样品测定
取盐酸米诺环素原位凝胶约2.5 g,置于100 mL量瓶中,加甲醇40 mL,充分振摇,超声助溶30 min,直至未见有胶状物质存于量瓶底部,加流动相稀释至刻度,摇匀,过滤,取续滤液作为供试品溶液,立即精密量取10 μL注入高效液相色谱仪,记录色谱图。4批样品的含量测定结果见表3。
2.7 讨论
在流动相的选择上,曾采用药典的0.2 mol·L-1醋酸铵-二甲基甲酰胺-四氢呋喃(600∶398∶2,内含0.2 mol·L-1乙二胺四乙酸二钠)为流动相,凝胶峰与样品峰有干扰 ,不能达到良好的分离度。而用乙酸铵缓冲盐溶液-乙腈(250∶ 105)为流动相时,保留时间适宜,峰形好,样品峰与凝胶峰无干扰。因此,本实验选择乙酸铵缓冲盐溶液-乙腈(250∶105)为流动相。
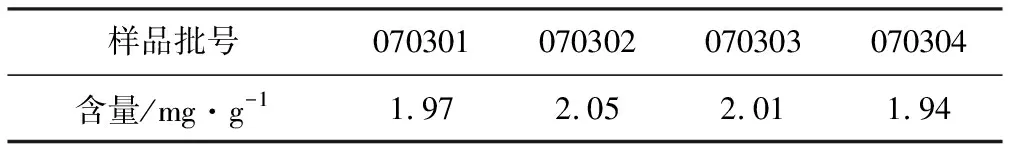
表3 样品含量测定结果(n=4)
3 结论
以乙酸铵缓冲盐溶液-乙腈(250∶105)为流动相,进样量10 μL,在350 nm处采用RP-HPLC法测定了盐酸米诺环素原位凝胶中盐酸米诺环素的含量,盐酸米诺环素浓度在50~1000 μg · mL-1(以米诺环素计)范围内与峰面积呈良好的线性关系。该方法操作简单,结果准确可靠,可用于盐酸米诺环素原位凝胶中盐酸米诺环素含量的测定。
[1] Baker P J,Evans R T,Slots J,et al. Susceptibility of human oral anaerobic bacteria to antibiotics suitable for topical use[J]. Journal of Clinical Periodontology,1985,12(3):201-208.
[2] 任蕾,杨圣辉,刘颖. 牙周炎常见菌对抗菌药物的筛选及活性测定[J]. 现代口腔医学杂志,2000,14(4):256-260.
[3] 国家药典委员会.中华人民共和国药典2005年版(二部)[S].北京:化学工业出版社,2005:697-698.
