脊髓小脑性共济失调3型的临床特征和基因诊断
2011-06-14王子峰李连平曲立贞李季春
王子峰,李连平,曲立贞,苏 净,李季春,曹 霞*
(1济宁医学院附属高唐县人民医院,山东高唐252800;2济南军区总医院)
脊髓小脑性共济失调是一种以肢体运动协调障碍为主要特点的常染色体显性遗传疾病。基因学检测是目前诊断遗传性脊髓小脑性共济失调(SCA)的金标准。近1年来,本研究对其临床特征及基因诊断方面的特征进行了探讨。现报告如下。
1 资料与方法
1.1 临床资料 收集临床诊断为SCA家系20例,均为汉族,其中患者10例、家系内正常人10例。家系1:先证者(Ⅲ6),男,38岁。该家系来自山东地区,家族无近亲结婚史。4代17例成员中女性患者2例,男性患者4例,发病年龄25~36岁,符合常染色体显性遗传规律。先证者体征:构音不清,偶有饮水呛咳,向右注视时可见慢眼动,向左注视时可见快速眼震。双上肢近端肌肉、双侧胸锁乳突肌明显萎缩,双上肢肌力Ⅴ级,双下肢肌力Ⅳ级。可见肌肉束颤。四肢肌张力增高。指鼻试验、双手轮替运动均不稳准,右侧重于左侧。双上肢肱二头肌反射、肱三头肌反射均活跃,双下肢膝腱反射、跟腱反射均亢进。髌阵挛、踝阵挛均阳性。双侧Hoffmans征阳性,双侧Babinski’s征阳性,余病理征未引出。脑膜刺激征阴性。家系内其他部分患者可见眼球内陷、双侧快速眼震等体征。家系2:先证者(Ⅲ6),女46岁,该家系来自山东地区,患者母亲、舅舅、1个弟弟均有类似症状,4代14例成员中女性患者2例,男性患者2例,最高发病年龄52岁,最低发病年龄42岁,平均发病年龄46岁,符合常染色体显性遗传规律。先证者体征:神志清楚,言语清晰,颅神经检查未发现异常,四肢肌力Ⅴ级,肌张力正常,闭目难立征(+),T6以下痛觉减退,双下肢末端触觉减退,双侧膝关节以下振动觉减退,双侧腱反射对称减弱,病理征未引出,颈软,脑膜刺激征阴性。家系1遗传家系图:见图1(使用中国遗传咨询网家系图在线绘制工具绘制)。家系2遗传家系图:见图2(使用中国遗传咨询网家系图在线绘制工具绘制)。

图1 家系1遗传家系图
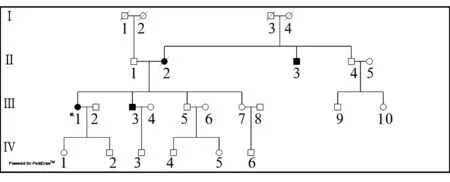
图2 家系2遗传家系图
1.2 实验方法
1.2.1 PCR反应 使用TaKaRa DNA提取试剂盒(大连宝生物工程有限公司提供)提取患者静脉血中DNA。SCA3亚型引物的PCR参照文献。2%琼脂糖凝胶电泳后紫外灯下观察PCR扩增产物,发现2个家系SCA3引物PCR扩增均发现异常条带,考虑为 SCA3亚型。使用 TaKaRa Agarose Gel DNA Purifiation Kit Ver.2.0琼脂糖凝胶DNA回收试剂盒(大连宝生物工程有限公司提供)回收DNA。
1.2.2 PCR产物克隆及测序 家系1先证者SCA3引物PCR产物电泳结果,可见400 bp大小异常条带,回收此条带DNA进行纯化。将已纯化PCR产物使用 TaKaRa DNA Ligation Kit Ver.2.0(Code No.D6022)中的SolutionⅠ直接和载体pMD18-T(Code No.D101A)连接,连接产物热转化至E.coli Competent Cell JM109中,涂布平板(LB液体培养基),37℃过夜培养,观察有无菌落。然后挑取不同白色单个菌落加入LB液体培养基中培养5 h以上,以此菌液为模板,使用M13-47/RV-M(M13-47碱基序列:CGC CAG GGT TTT CCC AGT CAC GAC;RV-M碱基序列:GAG CGG ATA ACA ATT TCA CAC AGG)引物进行PCR扩增,电泳检测。并同时移板(LB液体培养基),挑选阳性克隆子进行基因序列测定。将选定的含有阳性克隆的单菌落进行植菌,利用TaKaRa MiniBEST Plasmid Purification Kit Ver.2.0(Code No.DV801A)提取质粒。在 ABI PRISM 3130XL型全自动DNA测序仪上使用RV-M引物对质粒测序。
2 结果
2.1 部分患者琼脂糖凝胶电泳结果 家系1先证者及家系内其他患者,均可见一条约250 bp基因条带和一条400 bp异常基因条带。因等位基因一条来自父方,一条来自母方,家系1先证者为父系遗传,因此250 bp基因条带来自等位基因的母系基因,400 bp异常基因条带来自等位基因的另一条父系异常基因。家系1正常人SCA3 PCR产物均可见约250 bp正常扩增条带。先证者儿子PCR产物电泳结果,未见异常电泳条带。家系2患者SCA3 PCR扩增产物琼脂糖凝胶电泳均可见约400 bp异常条带。家系2正常人SCA3 PCR产物凝胶电泳扩增条带均可见约250 bp左右正常电泳条带,未见异常基因扩增带。
2.2 2个家系患者发病特点及部分测序结果 见表1。
3 讨论
脊髓小脑性共济失调3型是SCA中最为常见
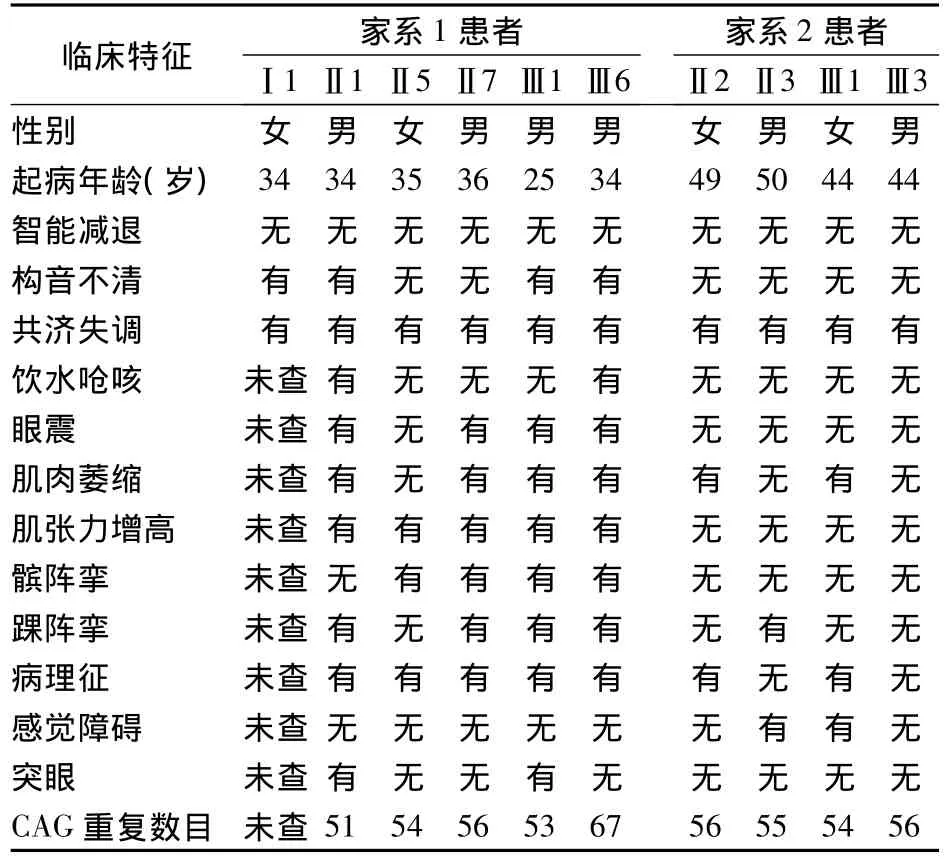
表1 2个家系患者发病特点及部分测序结果
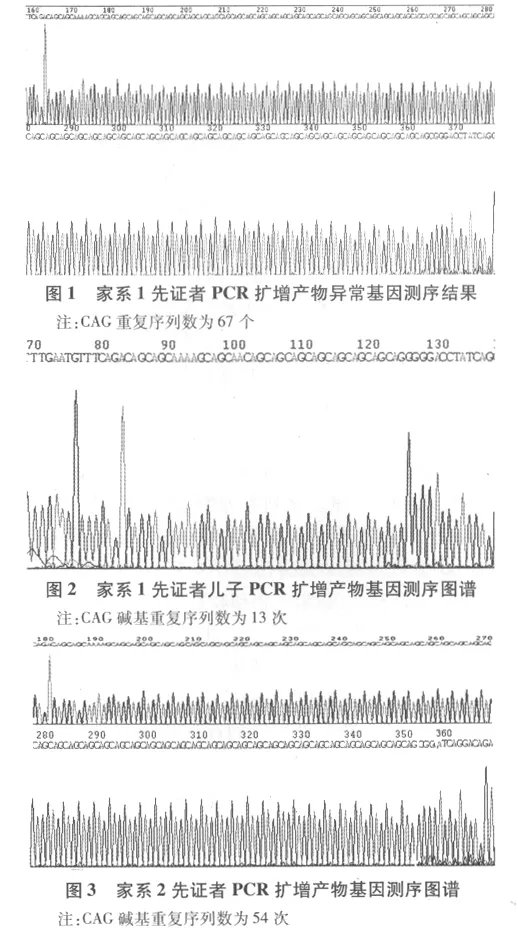
2.3 2个家系部分患者SCA3 PCR扩增产物基因测序 见图1、图2、图3。的类型,在汉族人群的脊髓小脑性共济失调中约占50%。SCA3的临床表型具有高度的遗传异质性和临床变异性,根据目前研究,大致分为5种不同临床亚型[2,3],根据此种分型,家系 1 患者病情严重,发病年龄较早,主要表现为小脑体征,同时伴有严重的锥体系和锥体外系症状,部分患者可见突眼,属于第1种亚型。家系2患者症状相对较轻,起病晚,伴有周围神经病变为其特点,属于第3种亚型。
关于SCA3亚型发病机制,研究表明系由突变的CAG重复碱基片段编码含有多聚谷氨酰胺片段的蛋白所致病[4]。正常人与患者的主要区别表现在CAG重复数目的不同,正常人CAG重复数目一般为12~41次,患者62~84次。国内谢秋幼报道,SCA3患者CAG重复数目为67~85次。本研究中的2个SCA3家系患者CAG重复数目为51~67次,明显低于谢秋幼等报道。且2个家系患者代间传递中有明显的发病年龄提前趋势,从表1可以看出家系1患者发病年龄第3代比第2代提前,症状有加重趋势;家系2患者发病年龄第3代比第2代也明显提前5岁左右,症状程度类似。家系1患者临床症状在代间传递中有明显的加重趋势。在此表中还可看出,2个家系CAG重复序列数在第3代患者中比第2代明显增多。这也提示临床症状、发病年龄均与CAG重复数目有关,CAG重复次数越多发病年龄越小且症状越重[4],进一步证明了此前提到的遗传早现现象。遗传早现现象主要在父系遗传中更明显。家系1先证者属于父系遗传,也说明了这一点。在家系2先证者属于母系遗传,家系2第3代患者中CAG重复序列数比第2代CAG序列数无明显增多,虽发病年龄提前,但症状无加重,需进一步搜集病例分析母系遗传中SCA3的表现。此外,家系1中Ⅲ1 CAG重复序列数为53次,Ⅲ6 CAG重复序列数为67次,但Ⅲ1患者发病年龄比Ⅲ6发病年龄提前9岁;家系2部分患者中CAG重复序列数多于家系1患者,但临床症状却较家系1患者轻,这种现象可能与遗传异质性有关。由于不同患者具有不同的遗传基础,从而导致遗传方式、病程进展、发病年龄、预后以及复发风险、病情严重程度等都可能不同。研究表明,遗传异质性的存在是导致遗传病病种增多的原因之一。
家系2中Ⅰ3、Ⅰ4生前均没有共济失调症状,死因也可排除SCA类疾病,但在子代中出现了发病患者。这是因为CAG重复片段所在的等位基因具有多态性的特点[5],如果等位基因携带有处于正常与异常之间的CAG重复数目(即所谓带有中间数目的CAG重复片段,国外报道SCA3一般为48~51次[6]),由于CAG重复片段的不稳定性,在下一代中CAG重复易发生新的突变发病,尤其在父系遗传中更明显。因此推测家系2的Ⅰ3(男性,75岁时去世)可能携带有中间数目的CAG重复片段。
SCA3症状前患者,是指基因检测CAG重复数目异常,尚无临床症状的人。由于SCA3是一种迟发性神经变性疾病,因此症状前患者达到一定年龄必定会出现症状。在目前尚无有效治疗方法的情况下,基因学检查明确症状前诊断防止该病发生具有重要意义。在本实验的家系1中,Ⅲ1属于症状前患者,因为在留取血样时,患者尚未出现临床症状,经基因检测CAG重复数目为53次,在随访2 a后患者出现共济失调症状,且发病年龄较其父亲提前,症状更重。这也证实了症状前基因诊断预测发病的准确性。症状前诊断的重要意义在于使SCA家系中正常人对于自己是否发病有准确的预测,使症状前患者在婚育年龄对下一代做到产前诊断,防止带有异常基因的患儿出生。对于经基因诊断明确为SCA3或其他SCA亚型的患者,对家系内正常人均应留取血液进行基因学检测,筛选家系内的症状前患者,一旦诊断CAG重复序列异常,即可诊断成立,其致病基因的外显率为100%。
[1]段晓慧,顾卫红,王国相.常染色体显性遗传脊髓小脑性共济失调表型和基因型的相关性[J].中日友好医院学报,2008,22:41-43.
[2]殷鑫浈,张宝荣,吴鼎文,等.脊髓小脑性共济失调第7型的临床特征及基因突变研究[J].遗传,2007,29(6):688-692.
[3]Dürr A,Stevanin G,Cancel G,et al.Spinocerebellar ataxia 3 and Machado-Joseph disease:clinical,molecular,and neuropathological feature[J].Ann Neurol,1996,39(4):490-499.
[4]Sakai T,Kawakami H.Machado-Joseph disease:a proposal of spastic paraplegic subtype[J].Neurology,1996,46(3):846-847.
[5]Ikeda H,Yamaguchi M,Sugai S.Expanded polyglutamine in the Maehado-Joseph Disease protein induces cell death in vitro and in vivo[J].Nat genet,1996,13(2):196-202.
[6]Viktor Honti,László Vécsei.Genetic and molecular aspects of spinocerebellar ataxias[J].Neuropsychiatric Disease and Treatment,2005,1(2):125-133.
