冬凌草分散片溶出度测定方法的研究
2011-05-26李其凤冯柏康李卫民
李其凤, 冯柏康, 李卫民, 肖 飞
(广州中医药大学,广东广州 510405)
冬凌草分散片是由冬凌草片经改剂型而来,原 剂型收载于《中华人民共和国卫生部药品标准》[1]中药成方制剂第3册,功能主治:清热消肿,用于急慢性扁桃体炎、咽炎、喉炎、口腔炎,试用于抗癌。大量的实验与文献证明[2-5],冬凌草中的有效活性成分为二萜类,其中以冬凌草甲素、冬凌草乙素抗癌作用最为突出。由于冬凌草甲素、冬凌草乙素在水中的溶解度极低,冬凌草片在服用后很难被身体消化吸收、发挥作用,因此开发其为适用于难溶性成分的分散片剂型。本文以HPLC法测定冬凌草甲素的含量[6-7],对冬凌草分散片的体外溶出度进行了研究,为冬凌草分散片的快崩速溶作用提供试验依据,也为进一步临床研究提供理论依据。
1 仪器与试药
1.1 仪器 ZRS-8G智能溶出试验仪(天津市天大天发科技有限公司 );Waters 2695泵(美国Waters);Waters 2996二极管阵列检测器(美国Waters);EMPOWER数据处理软件系统(美国Waters)。
1.2 试药 冬凌草分散片(自制,批号:090703、090707、090711)。
1.3 对照品 冬凌草甲素对照品(中国药品生物制品检定所)。
1.4 试剂 甲醇为色谱纯(德国默克),水为制备纯水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件 RP-18e(5 μm)LiChroCART 250-4色谱柱,200 mm ×4.6 mm,5 μm;流动相:甲醇-水(50∶50);检测波长:240 nm;按冬凌草甲素色谱峰计算,理论塔板数不低于3 500。
2.2 线性关系的考察 称取冬凌草甲素对照品适量,精密称定,加甲醇制成每1 mL含0.2 mg的溶液,精密量取1 mL至10 mL量瓶中,加0.1 mol/L的盐酸溶液定容至刻度,摇匀,微滤,精密吸取上述对照品溶液 1、2、5、10、15、20 μL,分别注入液相色谱仪,按上述色谱条件测定峰面积,以对照品进样量
X(μg)为横坐标、峰面积值Y为纵坐标,绘制标准曲线,结果冬凌草甲素对照品在0.019 64~0.392 8 μg范围内具有良好的线性关系,回归方程为:Y=1 375 484.31X-19 258.69,r=0.999 9。
2.3 系统适应性试验 取冬凌草甲素对照品溶液、供试品溶液各20 μL,注入高效液相色谱仪,记录色谱图,可见供试品中冬凌草甲素与其他组分能达到基线分离,见图1、2。
图1 冬凌草甲素对照品HPLC图
图2 供试品HPLC图
2.4 溶出方法的选择[8-9]
2.4.1 溶出方法的选择
诸暨市的检调对接工作不仅成效显著,也得到学界的肯定。有研究者指出,诸暨市人民调解组织介入刑事和解的探索表明,检察机关与人民调解组织相互发力、优势互补,凝聚成推动刑事和解内在推动力,不仅避免了检察机关在调解过程中既当裁判员又当运动员的尴尬局面,而且一定程度上缓解了检察机关的“人案矛盾”,提高了刑事诉讼效率。[7]
根据药典要求,溶出方法分为转篮法与桨法,取冬凌草分散片分别以转篮法与桨法进行溶出度的测定,溶出介质为0.1 mol/L的盐酸900 mL,转速为100转,分别于45 min取样,微滤,依法进样测定。结果见表1。转篮法测定的溶出度较桨法低,且相对标准平均偏差较大。
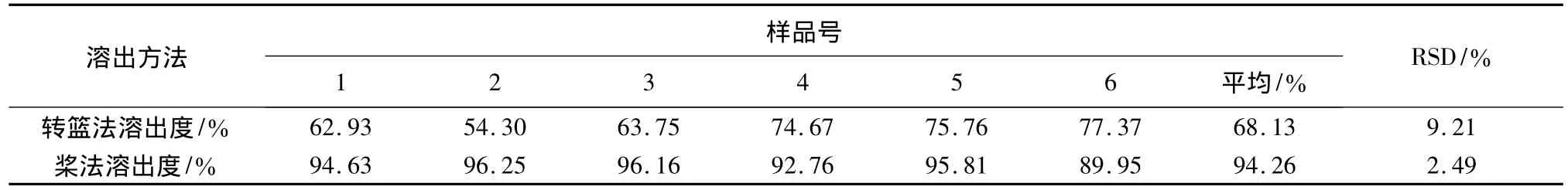
表1溶出方法的选择
结论:试验结果表明,选择《中国药典》2005年版二部附录X C溶出测定法第二法桨法作为溶出测定方法。
2.4.2 转速的选择
取冬凌草分散片按照《中国药典》2005年版二部附录X C溶出测定法第二法桨法分别以60、80、100、120 r/min进行溶出度的测定,溶出介质为0.1 mol/L的盐酸900 mL,分别于45 min取样,微滤,依法进样测定。结果见表2、图3。转速越大,溶出度越高,当转速达到100 r/min后,溶出度基本不再增加。选择100 r/min作为溶出测定的转速。测定,溶出介质为0.1 mol/L的盐酸900 mL,转速为100 r/min,分别于 5、10、15、30、45、60、90、120 min取样1 mL,微滤,依法进样测定。结果见表3。冬凌草分散片随时间的增加,溶出量亦相应增加,但在45 min后,溶出度增加很少,因此,确定溶出时间为45 min。
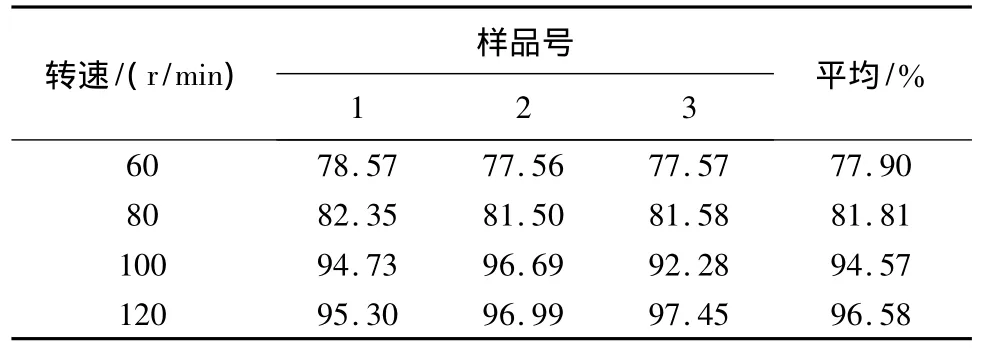
表2 不同转速下溶出度的比较/%
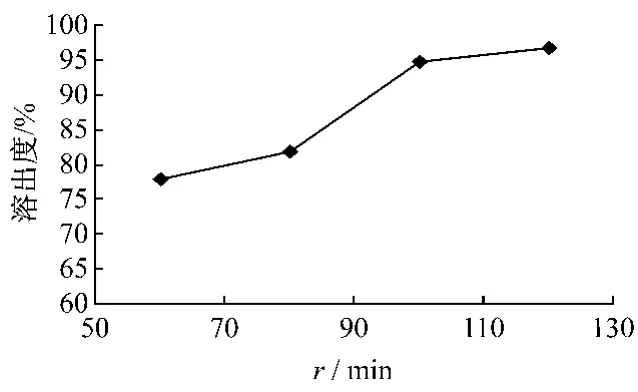
图3 不同转速下溶出度的比较
2.4.3 溶出曲线
取冬凌草分散片按照《中国药典》2005年版二部附录XC溶出测定法第二法桨法进行溶出度的
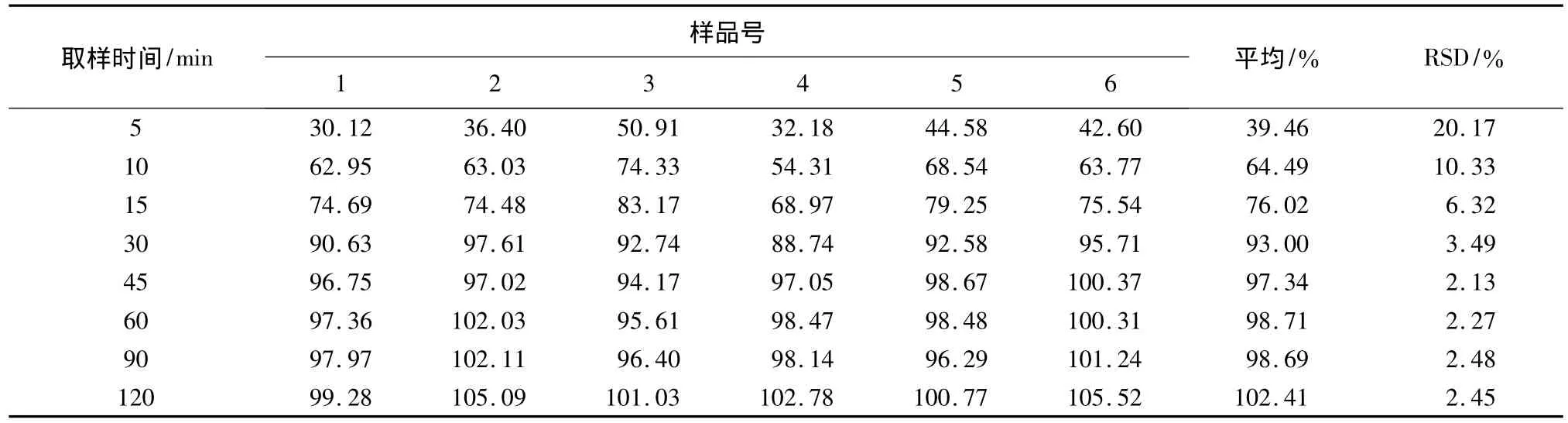
表3 不同取样时间的溶出度测定/%
2.5 空白试验 精密称取缺冬凌草的阴性样品,按溶出供试品溶液制备方法操作,依法进样测定,记录色谱图。结果在与冬凌草甲素对照品相同保留时间处无吸收峰。表明样品中无干扰冬凌草甲素的成分存在。
2.7 重复性试验 取一批样品,按上述溶出度测定方法,测定6次,计算溶出度,求得冬凌草甲素平均溶出度为96.10%,相对标准偏差RSD为1.14%。
2.8 稳定性试验 精密吸取供试品溶液,在室温下,分别于配制后 0、2、4、6、8、12 h 进行冬凌草甲素峰面积测定,每次进样10 μL,测得供试品中冬凌草甲素的平均峰面积值为238 214,RSD为1.99%。试验结果表明,供试品溶液在12 h内基本稳定。
2.9 回收率测定 取已知含量的同一批号样品,除去包衣,研细,取约16 mg,精密称定,精密加入冬凌草甲素对照品溶液(0.196 4 mg/mL)2 mL,置50 mL量瓶中,加入0.1 mol/L盐酸溶液约20 mL,超声处理(功率250 W,频率50 kHz)10 min,放冷,加0.1 mol/L盐酸溶液定容至刻度,摇匀,微滤,取续滤液。吸取10 μL,按色谱条件方法,进样,测定,计算,求得冬凌草甲素平均回收率为100.49%,RSD为1.73%。
2.10 样品溶出度测定 取本品,照溶出度测定法(《中国药典》2005年版二部附录X C第二法),以0.1 mol/L的盐酸溶液900 mL为溶出介质,转速为100 r/min,依法操作,经45 min时,取溶液5 mL,微滤,作为供试品溶液。另称取冬凌草甲素对照品适量,精密称定,加甲醇制成每1 mL含0.2 mg的溶液,精密量取 1 mL至 10 mL量瓶中,加 0.1 mol/L的盐酸溶液定容至刻度,摇匀,微滤,作为对照品溶液。吸取对照品溶液与供试品溶液各10 μL,进样测定,计算每片的溶出量。结果见表4。
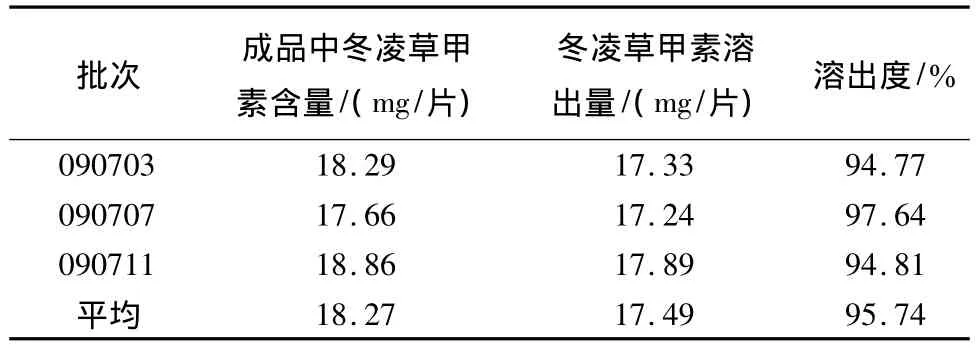
表4 样品含量测定
本品3批样品,平均溶出度为95.74%。
3 讨论
3.1 分别选用蒸馏水、0.1 mol/L HCl为溶出介质,试验结果表明以0.1 mol/L HCl为溶出介质的溶出度远远大于水,可能是由于冬凌草甲素在水中的溶解度极低。
3.2 分别以转篮法与桨法进行溶出度的测定,转篮法测定的溶出度较桨法低,且相对标准平均偏差较大。
3.3 采用桨法,以0.1 mol/L HCl为溶出介质,分别以60、80、100、120 r/min 进行溶出度的测定,结果显示转速越大,溶出度越高,当转速达到100 r/min后,溶出度基本不再增加。
3.4 分别于 5、10、15、30、45、60、90、120 min 进行溶出度的测定,结果显示冬凌草分散片随时间的增加,溶出量亦相应增加,但在45 min后,溶出度增加很少,因此,确定溶出时间为45 min。
3.5 制剂中其它成份对冬凌草甲素的溶出度测定无干扰,本方法较为准确可靠,可作为冬凌草分散片的质量控制的方法。
3.6 体外溶出试验结果表明,冬凌草分散片不仅崩解速度快,而且溶出迅速,为进一步的临床研究提供了理论依据。
[1]中华人民共和国卫生部.《卫生部药品标准》中药成方制剂[S].第3册.1991:59.
[2]刘晨江,赵志鸿.冬凌草的研究进展[J].中国药学杂志,1998,33(10):577-581.
[3]张典瑞,任天池.冬凌草甲素的药学研究进展[J].中国药学杂志,2003,38(11):817-820.
[4]李 钦,冯卫生.冬凌草化学成分、药理作用及开发研究进展[J].河南中医学院学报,2003,18(6):31-33.
[5]刘 净,谢 韬,魏秀丽,等.冬凌草化学成分的研究[J].中国天然药物,2004,2(5):276-279.
[6]金 珠,周丽莉.高效液相色谱法测定冬凌草中冬凌草甲素及冬凌草乙素的含量[J].时珍国医国药,3004,15(6):321-322.
[7]袁 珂,吴崇珍,张晓明,等.高效液相色谱法同时测定冬凌草中冬凌草甲、乙素的含量[J].中国现代应用药学杂志,2004,21(3):213-215.
[8]吴光辰,岳志伟.药物固体制剂的溶出度[M].北京:人民卫生出版社,1994:69.
[9]魏树理.生物药剂学与药物动力学[M].北京:北京医科大学和中国协和医科大学联合出版社,1997:179.
