溶胶-凝胶法制备钛酸锌微波介质陶瓷粉体影响因素的研究
2011-02-06余丽芳喻佑华袁玲玲李海南夏萌
余丽芳 喻佑华 袁玲玲 李海南 夏萌
(景德镇陶瓷学院,江西景德镇333001)
0 引言
微波介质陶瓷是指应用于微波频段(300MHz-300GHz)电路中作为介质材料并具有一定功能的陶瓷[1]。ZnO-TiO2系微波介质陶瓷因其本身具有较低的烧结温度(小于1100℃)及优良的微波介电性能而备受关注,但是,钛酸锌陶瓷中微波介电性能较好的六方钛铁矿型ZnTiO3相在950℃左右易发生分解反应,生成微波介电性能较差的尖晶石型Zn2TiO4相和金红石相,极大地影响陶瓷的介电性能[2]。
传统的固相反应法是在1000℃以上进行烧结的,偏钛酸锌极易分解,并且固相法制得的颗粒尺寸大,粒度分布宽,组分不均匀,粉体纯度不高以及化学活性不高,难以满足微波介质陶瓷越来越严格的质量要求。因此,作者采用溶胶-凝胶法制备偏钛酸锌粉体,主要探讨了钛/乙醇摩尔比(Ti/AE)、pH值、前驱体反应温度、预处理煅烧温度等对钛酸锌粉体的形成过程和性能的影响。
1 实验部分
实验所选用的原料均为分析纯试剂,所用的原料为:硝酸锌(Zn(NO3)2·6H2O)、钛酸四丁酯(Ti(OC4H9)4)、无水乙醇(CH3CH2OH,AE)、冰醋酸(CH3COOH)。
取物质量之比为n(Zn)∶n(Ti)=1∶1的硝酸锌和钛酸四丁酯,分别溶于适量乙醇中,然后将适当浓度的冰醋酸滴入钛酸四丁酯的乙醇溶液中,调节pH值在2~3的范围内,同时滴加硝酸锌,充分反应,将前驱体溶液充分水浴加热5h左右形成淡黄色透明溶胶,静置陈化12h后,溶胶转化成凝胶,将凝胶置于80℃下干燥24h,待完全变干后进行研磨、煅烧处理;然后进行造粒,在10MPa压力下压片,选择950~1050℃下煅烧2h(见图1)。
将预烧得到的粉体用德国Bruker AXS公司的D8 Advance型X射线衍射仪(XRD)进行物相分析,辐射源为
2 结果与讨论
2.1 钛酸四丁酯乙醇溶液的配制顺序与浓度控制
2.1.1 钛酸四丁酯乙醇溶液的配制顺序对溶胶制备的影响

图1 前驱体粉末合成工艺流程图Fig.1 The preparation process of precursors

表1 钛酸四丁酯乙醇溶液滴加顺序Tab.1 The adding sequence of C16H36O4Ti and alcohol
本实验采用的是分析纯的无水乙醇,但是还含有少量的水份,而钛酸四丁酯遇水极易水解,基于此特点,应将无水乙醇缓慢加入钛酸四丁酯中,并不断搅拌,这样配制才能得到澄清的黄色溶液。实验发现无水乙醇的用量主要影响胶凝时间,无水乙醇的用量大,会降低醇盐溶液的浓度,导致胶化时间变长,这是因为多余溶剂的挥发所需时间较长,较好的体积比为1∶8,成胶时间在5~7h左右。
钛酸四丁酯乙醇溶液的配制顺序对溶胶制备的影响如表1所示。
2.1.2 醇盐溶液浓度对溶胶制备的影响
无水乙醇的加入主要起着溶剂的作用,此外还有稀释的作用。随着乙醇用量的增加,溶液的浓度变稀,溶胶粘度降低,使得凝胶时间延长。因为凝胶网络的形成,需要一定的浓度。乙醇加入量太多,使钛酸四丁酯和硝酸锌的浓度降低,从而使得Ti(OH)x(OC4H9)y单体很难接触,交联成链的几率变小,导致缩聚反应的速度变慢。醇的浓度在Ti∶AE=7∶1~6∶1时比较适宜,浓度过小导致凝胶时间变长,并且延长了后续干燥时间,同时造成凝胶体内包含大量的乙醇,导致干燥时乙醇大量挥发,使凝胶体变得疏松多孔,影响热处理后粉体的质量。醇盐浓度过大则会导致胶体的局部聚沉,使凝胶的成份分布不均匀,严重影响合成钛酸锌粉体的质量。
笔者采用溶胶-凝胶法合成钛酸锌粉体,其溶胶过程主要是通过钛酸四丁酯的水解缩聚反应实现的。由于钛酸四丁酯的负电性烷氧基-OR,使金属离子Ti4+极易受到氢核攻击,遇到水后会立即发生水解和聚合反应,甚至出现Ti(OH)4沉淀,破坏溶胶体系的稳定性,将冰醋酸加入钛酸四丁酯乙醇溶液中,H+的攻击加速水解,并以配位形式取代钛酸四丁酯中的烷氧基,直接与钛相结合而形成水解配合物Ti(OH)x(OAc)y(其中x+y=4),这类配合物在水解和缩聚过程中很难被破坏,从而抑制了钛酸四丁酯的水解,进而能够得到稳定的溶胶。通过实验摸索得到整个反应的pH值控制在2~3左右。
2.3 溶胶反应温度
文献资料中阐述认为钛酸四丁酯搅拌时的温度要低,以避免由于热效应生成一些不期望的复杂化合物。溶胶的反应温度影响着溶胶质量,表2是在pH值为2~3时,30℃、35℃、40℃、50℃、60℃下,形成钛酸锌溶胶质量的分布情况。钛酸丁酯与硝酸锌的混合溶胶水解温度越高,溶胶粘度随时间增加得越快,溶胶越不稳定。温度升高,溶胶体系水解反应速率增大,同时胶粒动能增大,相互之间碰撞几率增大,缩聚反应加快,故溶胶粘度短时间内迅速增大。温度升高时,有机溶剂乙醇会部分挥发出来,使得参与水解缩聚反应的反应物浓度进一步增大,缩聚反应所得的聚合物浓度也随之增大,缩聚和聚沉更易进行,表现为溶胶粘度短时间内迅速增大。水解温度为20℃时水解缩聚反应发生的动力学条件不足,反应驱动力小,致使反应进行缓慢,这样也不利于超细粉体的制备。故溶胶水解温度应选择在35℃为宜。

表2 不同温度溶胶的质量Tab.2 Quality of the solutions obtained at different temperatures

图2 前驱体粉末在不同温度预烧4 h的X射线图谱Fig.2 XRD patterns of precursor powders calcined for 4h
2.4 凝胶的干燥与热处理
湿凝胶内含有大量溶剂和水,干燥凝胶的目的就是为了去除残余的、有机溶剂和水。笔者考虑到整个溶胶的形成是在醇溶剂的环境中形成的,因此在干燥温度为80℃的条件下处理湿凝胶;然后将得到的干凝胶研磨,选择700℃、750℃、800℃、900℃、1000℃进行热处理4h。图2是将ZnTiO3前驱体干凝胶分别在700℃、750℃、800℃、900℃、1000℃进行热处理4h后粉体的X射线衍射图谱。在700℃热处理4h后,由XRD图谱可看出有ZnTiO3晶相的存在,并伴有少量的Zn2TiO4和TiO2,峰强都不够大。在800℃热处理4h后,明显看出该衍射峰强度增大,主晶相为ZnTiO3晶相,伴随有少量的Zn2TiO4和TiO2相存在,温度升高到900℃时,可以清楚地看出,ZnTiO3相的衍射峰值强度下降,取而代之的是Zn2TiO4相与TiO2相大量产生,当温度升高至1000℃热处理4h后,主晶相转变成Zn2TiO4晶相,并有大量TiO2相的存在,产生主晶相转变成Zn2TiO4的主要原因[3]是:

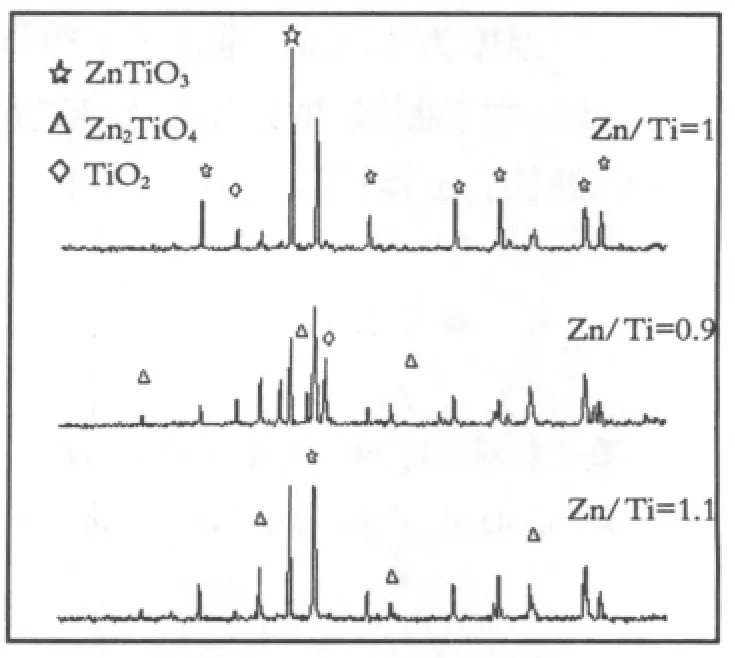
图3 不同T i/Z n比在800℃煅烧粉体的X射线图谱Fig.3 XRD patterns of powders calcined at 800℃with different Zn/Ti ratios
随着热处理温度的升高,Zn2TiO4晶相的衍射峰强度不断增大,结晶化程度不断提高,但当高于900℃时,ZnTiO3晶相开始分解,产生大量的Zn2TiO4和TiO2相。因此,对ZnTiO3干凝胶的预处理温度控制在800℃最佳,能得到较纯的ZnTiO3粉体。
2.5 不同i摩尔之比的物相分析
图3所示为将Zn/Ti分别为1.1,1.0,0.9的干凝胶粉末在800℃进行热处理4h后粉体的XRD分析。在Zn/Ti比为0.9时,衍射峰的杂峰很多,主晶相为偏钛酸锌相,但是衍射峰强度不够大,存在较多的金红石相与正钛酸锌相;在Zn/Ti比为1时,存在偏钛酸锌相,少量的正钛酸锌相,主晶相偏钛酸锌相较Zn /Ti比为1.1时,衍射峰强度更大,当Zn/Ti比增大至1.1时,正钛酸锌相的衍射峰强度增大,可能是发生了如下相转变反应[4]:ZnTiO3+ZnO→Zn2TiO4。
综合以上的分析可以得出:在800℃预烧温度下,粉料中的ZnTiO3相的衍射峰强度随着Zn2+离子摩尔量的增加而增大,当Zn/Ti=1时,此时的ZnTiO3相的衍射峰强度最大,继续增加Zn2+离子的摩尔量,ZnTiO3相的衍射峰强度反而变小,Zn2TiO4的衍射峰强度增大,这也意味着Zn/Ti=1是合成六方钛铁矿型ZnTiO3相的最佳的摩尔比。
3 结论
本文通过采用sol-gel法,以钛酸四丁酯Ti(OC4H9)4、低成本无机盐Zn(NO3)2·6H2O为原理,无水乙醇为溶剂,冰醋酸调节pH值制备ZnTiO3粉体通过试验分析得到最佳的工艺条件:溶液pH值为2~3,Ti∶AE=7: 1~6∶1,溶胶反应温度为35℃时,是合成前驱体粉料的最佳配方。并且六方钛铁矿型ZnTiO3粉末的最佳合成温度为800℃,最佳合成钛锌摩尔比为1∶1。
1何进.微波介质陶瓷材料综述.电子元件与材料,1995:7~14
2 DULIN F H,RASE D E.Phase equilibriums in the system ZnO-TiO2.J.Am.Ceram.Soc.,1960,43:125~131
3 CHAI YIN-LAI,CHANG YEE-SHIN,et al.Effect of borosilicate glass addition on the structure and dielectric properties of ZnTiO3ceramics.Mater.Res.Bull.,2008,43(2): 257~263
4 CHANG Y S.Synthesis and characterization of zinc titanate nano-crystal powders by sol-gel technique.J.Cryst.Growth, 2002,243:319~326.
