ZSM-5沸石的原位晶化及其石脑油催化裂解性能
2011-01-10陈希强汪哲明肖景娴
陈希强,汪哲明,肖景娴
(中国石油化工股份有限公司上海石油化工研究院,上海 201208)
ZSM-5沸石的原位晶化及其石脑油催化裂解性能
陈希强,汪哲明,肖景娴
(中国石油化工股份有限公司上海石油化工研究院,上海 201208)
采用 X射线粉末衍射(XRD)和扫描电镜(SEM)手段研究了合成条件对高岭土微球上原位晶化 ZSM-5沸石的影响,并以含C5~C12烃的石脑油为原料,在固定流化床上评价了不同催化剂的催化裂解性能。结果发现,采用转动晶化方式,控制合适的原料组成与晶化时间,能得到结构完整的含纯相ZSM-5沸石的高岭土微球。对比催化剂水热老化前后的XRD,氨气程序升温脱附(NH3-TPD)和 N2吸附-脱附以及石脑油催化裂解结果,发现磷元素改性能有效提高催化剂的水热稳定性,而且原位法制备的催化剂比半合成法制备的催化剂具有更高的水热稳定性。在650 ℃,常压,进料水油质量比1:1,质量空速1 h-1的条件下,在老化后的原位型催化剂作用下,石脑油的转化率达41.0%,乙烯和丙烯总收率达21.0%,均高于相同条件下半合成型催化剂的反应结果。
分子筛 高岭土微球 原位晶化 石脑油 催化裂解
乙烯和丙烯是两种重要的有机化工原料,主要通过石脑油非催化的蒸汽热裂解生产,但蒸汽热裂解反应温度高(800~880 ℃),能耗高,双烯(乙烯和丙烯)收率低,丙烯乙烯比低(0.4~0.6),而采用催化裂解石脑油生产乙烯和丙烯具有反应温度低(650~780 ℃),能耗低,双烯收率高及丙烯和乙烯比高(0.6~1.3)的优势[1]。常见的裂解催化剂可分为金属氧化物型[2]和分子筛型[3]两类,其中分子筛型催化剂因其强酸性与孔道择形性,可以进一步降低反应温度及灵活调变产物组成,因而成为当前研究的热点。催化裂解石脑油生产乙烯和丙烯是一个高温酸催化反应,因此催化剂极易积炭而失活,需采用流化床工艺实现其快速再生及循环,同时为了抑制积炭以及推动反应平衡朝生成烯烃的方向移动,需要在反应体系中引入水蒸气[4]。为此,催化剂除了需具备较高反应活性外,还需具备良好的水热稳定性。当前的流化床催化剂根据制备方法不同,可分为半合成型催化剂和原位型催化剂。其中,直接将分子筛、高岭土和粘结剂等组分与水混合打浆,然后喷雾干燥成型制得的为半合成型催化剂。该类型催化剂具有各组分分布不均匀,孔道易堵塞以及分子筛的利用率较低的缺点。而原位型催化剂是通过先将高岭土及粘结剂等组分喷雾成型,然后再进行晶化,将高岭土微球中的部分高岭土及粘结剂转化为分子筛,使得催化剂具有畅通的孔道结构,不但提高了分子筛的利用率,而且催化剂的反应活性、水热稳定性和抗重金属等性能也得到了增强[5]。目前,原位晶化Y型沸石的研究较多,且已有应用于工业流化催化裂化(FCC)的报道[6,7],而对于原位晶化ZSM-5沸石的研究相对较少[8],亦未见其应用于石脑油催化裂解的报道。本工作重点考察了 ZSM-5沸石原位晶化过程中原料组成、晶化方式及晶化时间对原位晶化的影响,并以含C5~C12烃的石脑油为原料,在固定流化床上评价了催化剂的催化裂解性能。
1 实验部分
1.1 实验方法
高岭土微球原位合成ZSM-5沸石。将高岭土(苏州阳山高岭土)、硅溶胶、水及少量晶种混合均匀,利用高速剪切机乳化后经喷雾干燥成型,制得粒径为20~100 μm的高岭土微球。将高岭土微球焙烧活化后与氢氧化钠、乙二胺、硅溶胶和水按比例混合,转移到不锈钢晶化反应釜中,于 170 ℃自生压力条件下晶化一定时间,取出后经过滤、洗涤、干燥和焙烧制得原位合成样品。
原位合成催化剂的制备。将原位合成样品置于5%的硝酸铵溶液中,于90 ℃条件下交换2 h,然后过滤、洗涤,重复上述过程两次后将样品干燥、焙烧,最后用磷元素修饰后得到原位合成催化剂,记作Situ-P,Situ-P经过650 ℃水蒸气老化2 h后记作Situ-PH。
半合成催化剂的制备。将所需量的高岭土、H-ZSM-5沸石、硅溶胶和水混合均匀,乳化后喷雾干燥成型,制得粒径为20~100 μm的高岭土微球。微球经过600 ℃焙烧后制得石脑油裂解催化剂,记作Semi。Semi经过650 ℃水蒸气老化2 h后记做Semi-H,Semi用磷元素修饰后记作Semi-P,Semi-P经过650 ℃水蒸气老化2 h后记做Semi-PH。其中半合成催化剂与原位合成所用的高岭土微球中的高岭土含量相同。
1.2 样品表征
样品的晶相采用日本理学D/max-1400型X射线粉末衍射(XRD)仪测定,工作电压30 kV,电流30 mA,扫描范围5~50°,并以2θ为7.9,8.7,23.0和23.8°处的ZSM-5的特征衍射峰峰高之和计算ZSM-5的相对结晶度。样品的形貌和颗粒尺寸采用日本JSM-35C型扫描电镜(SEM)观察。采用氨气程序升温脱附(NH3-TPD)测催化剂的酸性,样品量为100 mg,脱附温度从100 ℃升到600 ℃,升温速率10 ℃/min,热导检测器。样品的比表面和孔容采用Micrometrics公司Tristar3000型比表面分析仪测定。
1.3 石脑油催化裂解反应性能评价
催化剂评价在固定流化床上进行,原料为高桥石化提供的含C5~C12烃的石脑油,催化剂装填量130 g,石脑油与预热后的水蒸气同时通入反应器。评价条件:反应温度650 ℃,常压,水油质量比1:1,质量空速1 h-1。所得产物经冷阱冷凝后分离,气体产物用排水法收集并测其体积,用6890N双通道双检测器气相色谱仪分析其组成,其中烃类气体的分析采用HP PLOT/Al2O3型毛细管色谱柱,FID检测器,非烃类气体的分析采用13X分子筛色谱柱,热导检测器。石脑油的转化率以生成气态的C1~C4烃类与氢气的质量之和除以石脑油的进料量计算得到,乙烯和丙烯的收率以收集到的乙烯和丙烯的质量分别除以石脑油进料量计算得到,选择性由其相应的收率除以石脑油转化率得到。
2 结果与讨论
2.1 高岭土微球上原位晶化ZSM-5沸石
2.1.1 高岭土活化

图1 经过不同温度焙烧的高岭土XRD图谱Fig.1 XRD patterns of Kaolin calcinated at different temperature
未焙烧的高岭土中硅和铝物种以晶体的形式存在,性质稳定,不易与酸碱反应,因此在晶化之前需将高岭土活化,使其中的硅和铝易生成沸石晶体。其中高温焙烧就是一种比较常见的高岭土活化方法。图1是不同温度焙烧后高岭土的XRD谱图。从图中可以看出,焙烧之前高岭土具有很强的晶体衍射峰,而经过600~900 ℃的高温焙烧,样品变成了无定形的偏高岭土,而继续升高焙烧温度到1 000 ℃甚至更高时,又在2θ为46.5º附近出现了新的衍射峰,说明有新的晶相生成。由于升高焙烧温度,能提高高岭土中活性硅铝物种的比例,而ZSM-5沸石恰恰是一种高硅铝比的沸石,因此本实验选用900~1 000 ℃的焙烧温度来活化高岭土。
2.1.2 原料组成对原位晶化的影响
以高岭土微球的质量为基准,考察了氢氧化钠和水的加入量对原位晶化的影响,结果如表1所示。从表中可以看出,固定水的用量,随着体系中氢氧化钠加入量的增加,产物由无定形物质逐渐转变为ZSM-5沸石,继续增加碱量,出现P型沸石等杂晶。这是由于高碱度有利于富铝沸石的形成[9],而P型沸石的硅铝比相比ZSM-5沸石低很多,因而提高体系碱度,产物由ZSM-5沸石向P沸石转化。固定氢氧化钠的加入量,在氢氧化钠与高岭土微球质量比为0.08时,随着水量的增加,产物逐渐由ZSM-5沸石转变为无定形物质。这说明要在高岭土微球上原位合成纯相的ZSM-5沸石,必须控制合适的碱量和水量。

表1 原料组成对晶化的影响Table 1 Effect of raw material composition on crystallization

图2 采用不同晶化方式原位合成样品的XRD图谱Fig.2 XRD patterns of in-situ samples synthesized by different crystallization modes
2.1.3 晶化方式对原位晶化的影响
图2 是不同晶化方式下原位合成样品的 XRD谱图。对比标准XRD谱图可知,动态条件下(搅拌和转动)得到的是纯相ZSM-5沸石,而静态条件下,产物中除了ZSM-5沸石,还混有P型沸石。由于高岭土微球的原位晶化是一个硅铝物种凝胶化和凝胶组装为沸石的平衡过程[10],而动态晶化可有效强化液固传质,保障晶化体系的均匀性,因此有利于抑制杂晶生成。
图3是不同晶化方式下原位合成样品的扫描电镜照片。从照片上看,静态条件下(图3a)微球表面不太平整,可能与晶化过程中沸石生长不均匀,与液固传质不佳有关。而搅拌条件下(图3b)微球破损较严重,可能是搅拌过程中微球与微球以及微球与搅拌浆之间的碰撞或摩擦太强烈所致。采用转动晶化釜的晶化条件下(图3c),微球形貌保持较好,表面也比较光滑,可以看到微球表面均匀分布着大小为0.2~0.8 μm的棒状结晶。结合XRD和SEM结果可知,转动晶化既能抑制杂晶的生成,又能保证高岭土微球的完整性,不影响其实际应用,是一种较适宜的原位晶化方式。

图3 不同晶化方式合成样品的SEM结果Fig.3 SEM images of in-situ samples synthesized by different crystallization modes
2.1.4 晶化时间对原位晶化的影响
图4给出的是高岭土微球原位晶化ZSM-5沸石的晶化曲线。可以看出,该曲线呈现出典型的S型,与一般的沸石合成过程类似。在晶化4 h之前,样品的结晶度小于10%,并且随着晶化时间延长缓慢增加,此阶段可认为是诱导成核期;4 h之后,样品的结晶度随着晶化时间延长迅速增加,并在20~24 h达到最大,此阶段是晶体生长期。继续延长晶化时间,ZSM-5的结晶度稍有下降,这可能是碱性条件下,部分ZSM-5晶体溶解并重新组装所致[11],因而在本实验体系下适宜的晶化时间为20~24 h。

图4 ZSM-5沸石相对结晶度随晶化时间的变化Fig.4 Relative crystallinity of in-situ ZSM-5 vs crystallization time

图5 不同催化剂的XRD图谱Fig.5 XRD patterns of different catalysts
2.2 石脑油裂解催化剂表征
图5为不同催化剂的XRD谱图。从图中可以看出,Semi催化剂经过水热老化后,其结晶度下降较明显,而Semi-PH和Situ-PH的结晶度变化不明显。这说明磷元素修饰能有效提高ZSM-5沸石的骨架稳定性,减少了水热脱铝的程度。对比Semi-P和Situ-P的XRD结果,发现两者的结晶度相差不大,因此,也可以认为 Situ-P中的 ZSM-5沸石含量与 Semi-P中的ZSM-5沸石含量接近。

图6 不同催化剂的NH3-TPD曲线Fig.6 NH3-TPD curves of different catalysts
图6是不同催化剂的NH3-TPD表征。其中脱附温度150~310 ℃的峰代表弱酸位,主要由沸石的表面硅羟基产生;310~500 ℃的峰代表强酸位,主要由沸石的桥羟基Si(OH)Al产生[12],与骨架铝的含量密切相关。可以看出,未经磷元素修饰的催化剂Semi经过水热老化后,酸量下降非常明显,而经过磷修饰的Semi-P催化剂,下降程度则较小,采用原位法制备的催化剂Situ-P经过水热老化后,虽然弱酸酸量依然有明显下降,但是强酸酸量下降得非常少。进一步利用 Gaussian函数分峰拟合计算强酸和弱酸位的峰面积,结果列于表2。

表2 不同催化剂的酸量及分布Table 2 Acid amount and distribution of different catalysts
从表2中可以更直观地看出,未修饰的催化剂Semi经水热老化后,总酸量的保留率为50.2%。磷修饰的半合成催化剂Semi-P经水热老化后,总酸量从86.4%下降到53.9%,保留率提高到62.4%,这说明磷元素的引入能有效地提高催化剂中沸石骨架的水热稳定性。磷修饰的原位型催化剂Situ-P经水热老化后,总酸量从74.2%下降到64.7%,保留率高达87.2%,这说明采用原位晶化法制备的催化剂比采用半合成法制备的催化剂具有更高的水热稳定性。
表3列出的是不同催化剂的N2吸附-脱附结果。可以发现,其中任何一种催化剂经过水蒸气老化之后,其比表面和孔体积都有不同程度的下降,而平均孔径则有所增加,与郑淑琴等[7]在研究原位 FCC催化剂时所得结果类似。这可能是因为高温水蒸气引起沸石骨架脱铝[3],而脱出的非骨架铝继续堆积在沸石孔道内,从而降低沸石的比表面和孔体积。对比催化剂水热前后的数据还可以发现,催化剂的微孔比表面积比总比表面积下降更明显,如Semi-P催化剂,水热后微孔比表面积下降了7.8 m2/g,但是催化剂总比表面积只下降了6.5 m2/g。这表明催化剂中介孔或者大孔的比表面积经过水蒸气老化后有所增加,可能是因为高温水蒸气具有疏通催化剂中介孔或者大孔孔道的作用。
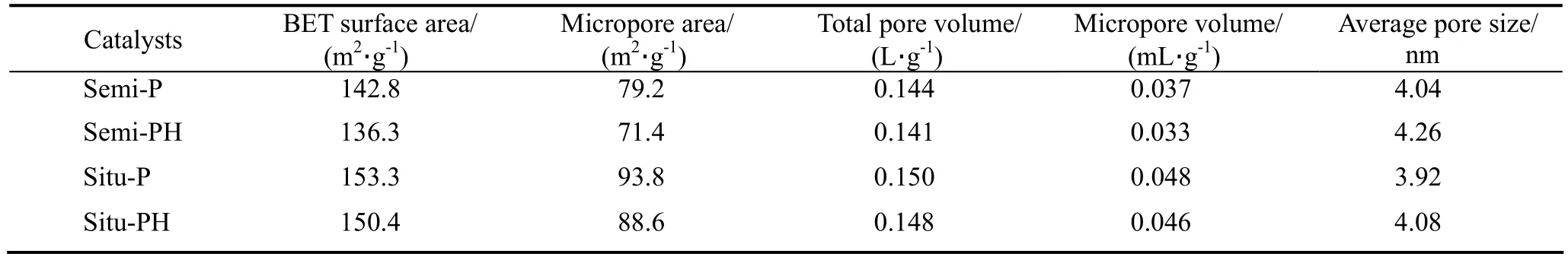
表3 不同催化剂的比表面积和孔体积Table 3 Surface area and pore volume of different catalysts
结合XRD,NH3-TPD和N2吸附-脱附结果,可以认为原位晶化法制备的催化剂具有较高的水热稳定性。可能的原因是晶化过程中部分高岭土基质及粘结剂被转化成沸石,从而使催化剂具有畅通的孔道结构,有利于水蒸气扩散,减少了水蒸气对沸石晶体的冲击和破坏,而且由于原位生长的ZSM-5沸石与高岭土基质之间的结合力较强,从而使原位型催化剂具有较高的水热稳定性。
2.3 石脑油催化裂解性能评价
选择4种经过磷修饰的催化剂,考察其催化裂解性能,结果列于表4。从石脑油的转化率看,老化之前,半合成型催化剂Semi-P比原位型催化剂Situ-P活性更高,这与其具有较多的酸量有关。经过老化后,半合成型催化剂Semi-PH的活性下降明显,石脑油转化率从45.6%下降到37.7%,保留率为82.7%,而原位型催化剂上石脑油的转化率则从43.0%下降到41.0%,保留率达到95.3%,可见,原位型催化剂比半合成催化剂具有更佳的水热稳定性,与之前N2吸附-脱附及NH3-TPD表征结果一致。
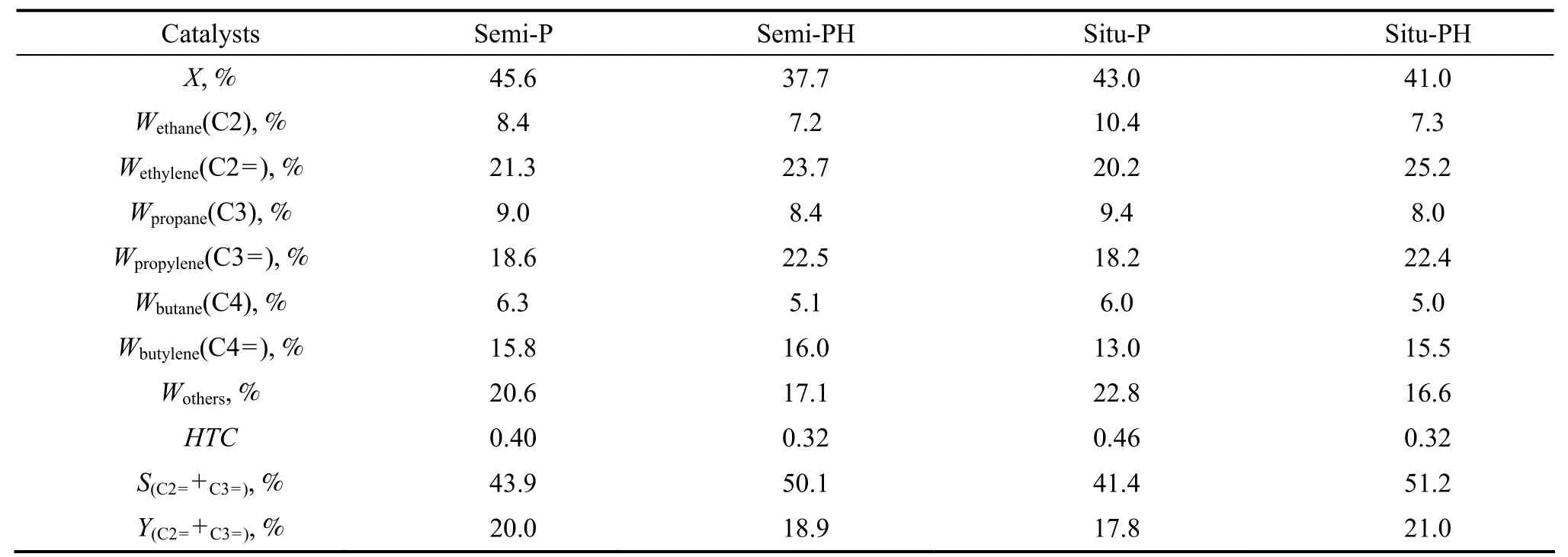
表4 不同催化剂的石脑油催化裂解结果Table 4 The results of naphtha catalytic cracking over different catalysts
气相产物的分析显示,催化剂经过老化后,产物中烷烃选择性下降,烯烃选择性上升,目标产物乙烯和丙烯的选择性也明显增加。这是因为水热老化降低了催化剂的酸量,而较低的酸量能够降低双分子反应的几率,抑制氢转移反应[13],导致烷烃的选择性下降,这从表 4中代表氢转移反应活性的氢转移指数(HTC)[14]下降也得到证实。
从表4还可以看出,经过水热老化后,Semi-PH和Situ-PH两种催化剂的乙烯与丙烯的选择性之和相近,都在50%以上,但是因为Situ-PH的反应活性比Semi-PH更高,因此,其最终目标产物中乙烯和丙烯的收率之和更高,其中丙烯和乙烯比可达0.89,高于传统的蒸气裂解技术(丙烯和乙烯比为0.4~0.6)。
3 结 论
a)在 170 ℃,氢氧化钠与高岭土微球质量比为 0.08~0.12,水与高岭土微球质量比为 4~10,水热晶化20~24 h的条件下,在高岭土微球上原位合成了纯相ZSM-5沸石。转动晶化在保证微球完整性的同时能有效强化液固传质,抑制杂晶生成,是一种较好的原位晶化方式。
b)采用磷元素修饰催化剂能有效提高催化剂的水热稳定性;原位法制备的催化剂比半合成法制备的催化剂具有更高的水热稳定性。
c)在650 ℃,常压,进料水油质量比1:1,质量空速1 h-1的反应条件下,在老化后的原位型催化剂上石脑油转化率41.0%,乙烯和丙烯总收率21.0%,丙烯和乙烯比为0.89。
[1]刘学龙, 鲁建春, 周春艳, 等. 石脑油催化裂解制烯烃技术经济分析[J]. 石油化工, 2005, 34(增刊): 80-81.Liu Xuelong, Lu Jianchun, Zhou Chunyan, et al. Techno-economy analysis of naphtha catalytic cracking for olefin production[J]. Petrochem Technol, 2005, 34(Supplement): 80-81.
[2]李小明, 宋芙蓉. 催化裂解制烯烃的技术进展[J]. 石油化工, 2002, 31(7): 569-573.Li Xiaoming, Song Furong. Advances in olef in production technology by catalytic cracking[J]. Petrochem Technol, 2002, 31(7): 569-573.
[3]Wan J L, Wei Y X, Liu Z M, et al. A ZSM-5-based catalyst for efficient production of light olefins and aromatics from fluidized-bed naphtha catalytic cracking[J]. Catalyis Letters, 2008, 124: 150-156.
[4]汤效平, 周华群, 魏 飞, 等. 催化裂解多产丙烯过程热力学分析[J]. 石油学报(石油加工), 2008, 24(1): 22-27.Tang Xiaoping, Zhou Huaqun, Wei Fei, et al. Thermodynamic analysis of propylene-enhancing FCC process[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2008, 24(1): 22-27.
[5]王有和, 李 翔, 刘欣梅, 等. 高岭土微球上无胺法ZSM-5的原位合成[J]. 无机化学学报, 2009, 25(3): 533-538.Wang Youhe, Li Xiang, Liu Xinmei, et al. In-situ synthesis of ZSM-5 on Kaolin microspheres from amine-free system[J]. Chinese Journal of Inorganic Chemistry, 2009, 25(3): 533-538.
[6]Brown S M, Durante V A. Fluid catalytic cracking catalyst comprising microspheres containing more than about 40 percent by weight Y-Faujasite and methods for making: US, 4493902[P]. 1985-1-15.
[7]郑淑琴, 蒋文庆, 索继栓. 高岭土型FCC催化剂的特性研究[J]. 工业催化, 2003, 11(5): 49-52.Zheng Shuqin, Jiang Wenqing, Suo Jiquan. Study on the properties of kaolin-based FCC catalysts[J]. Industrial Catalysis, 2003, 11(5): 49-52.
[8]孙书红, 马建泰, 庞新梅, 等. 高岭土微球合成ZSM-5沸石及其催化裂化性能[J]. 硅酸盐学报, 2006, 34(6): 757-761.Sun Shuhong, Ma Jiantai, Pang Xinmei, et al. Synthesis of ZSM-5 in Kaolin microspheres and it’s fluidized catalytic cracking performance[J].Journal of the Chinese Ceramic Society, 2006, 34(6): 757-761.
[9]徐如人, 庞文琴等. 分子筛与多孔材料化学[M]. 北京: 科学出版社, 2004: 212-219.
[10]郑淑琴, 谭争国, 丁 伟, 等. 高岭土原位合成Y型沸石过程中的吸附研究[J]. 无机盐工业, 2006, 38(1): 62-64.Zheng Shuqin, Tan Zhengguo, Ding Wei, et al. Study on synthesis of zeolite Y from Kaolin in-situ technology by adsorption method[J].Inorganic Chemicals Industry, 2006, 38(1): 62-64.
[11]冯 会, 李春义, 山红红. 晶化时间对高岭土微球上ZSM-5沸石的原位合成及其催化性能的影响[J]. 石油学报(石油加工), 2008, 24(4):438-445.Feng Hui, Li Chunyi, Shan Honghong. Effect of crystallization time on in-situ synthesis and catalytic properties of ZSM-5 zeolite on Kaolin microspheres[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2008, 24(4): 438-445.
[12]Topsøe N Y, Pedersen K, Derouane G E. Infrared and temperature-programmed desorption study of the acidic properties of ZSM-5-type zeolites[J]. Journal of Catalysis, 1981, 70(1): 41-52.
[13]朱根权, 张久顺, 汪燮卿. 丁烯催化裂解制取丙烯及乙烯的研究[J]. 石油炼制与化工, 2005, 36(2): 33-37.Zhu Genquan, Zhang Jiushun, Wang Xieqing. Study on the production of propylene and ethylene by catalytic cracking of butylenes[J].Petroleum Processing and Petrochemicals, 2005, 36(2): 33-37.
[14]沈志虹, 付玉梅, 蒋 明, 等. 化学改性对催化裂化催化剂氢转移性能的影响[J]. 催化学报, 2004, 25(3): 227-230.Shen Zhihong, Fu Yumei, Jiang Ming, et al. Effects of chemical modification on hydrogen transfer activity of cracking catalyst Y[J]. Chinese Journal of Catalysis, 2004, 25(3): 227-230.
In-situ Crystallization of ZSM-5 Zeolite and Catalytic Cracking Performance for Naphtha
Chen Xiqiang, Wang Zheming, Xiao Jingxian
(Shanghai Research Institute of Petrochemical Technology, China Petrochemical Corporation, Shanghai 201208, China)
The effects of synthesis conditions on in-situ crystallization of ZSM-5 zeolite on Kaolin microspheres were investigated by means of X-ray diffraction(XRD) and scanning electron microscope(SEM). The catalytic cracking properties of the catalysts for naphtha with C5-C12 hydrocarbon were also investigated in a fixed fluidized bed. The results showed that integrated Kaolin microspheres with pure ZSM-5 zeolite could be prepared by rotary crystallization mode through adjusting material composition and crystallization time. The comparison of XRD, temperature-programmed desorption(NH3-TPD), N2adsorption-desorption and naphtha cracking results of catalysts before and after steam aged indicated that the phosphorus modification effectively improved the hydrothermal stability of catalysts and the in-situ catalyst exhibited superior hydrothermal stability to semi-synthesized catalyst. Under the conditions of 650 ℃, mass hourly space velocity(WHSV) 1.0 h-1and mass ratio of water to oil 1.0, high conversion of naphtha (41.0%) and high yield of ethylene-plus-propylene (21.0%)were achieved over the steam aged in-situ catalyst.
zeolite; Kaolin microspheres; in-situ crystallization; naphtha; catalytic cracking
O643.32;TQ426.95 文献标识码:A
1001—7631 ( 2011 ) 06—0481—07
2011-11-10;
2011-11-30
陈希强(1982-),男,工程师;汪哲明(1976-),男,高级工程师,通讯联系人。E-mail: zmwangdicp@126.com
国家自然科学基金石油化工联合基金资助项目(20736011)
