海水中文石溶解、沉淀速率及动力学研究
2010-12-28陶小晚罗春树陈利新王先超
陶小晚,罗春树,陈利新,王先超
(1. 中国石油勘探开发研究院,北京 100083;2. 中国石油塔里木油田分公司勘探开发研究院,新疆 库尔勒 841000;3. 山东中煤物探测量总公司,山东 泰安 271000)
海水中文石溶解、沉淀速率及动力学研究
陶小晚1,罗春树2,陈利新2,王先超3
(1. 中国石油勘探开发研究院,北京 100083;2. 中国石油塔里木油田分公司勘探开发研究院,新疆 库尔勒 841000;3. 山东中煤物探测量总公司,山东 泰安 271000)
利用free-drift开放反应系统,在恒压力 (1×105Pa)、恒温度 (25.0 ℃ ± 0.2 ℃) 环境下,研究人工海水中近溶解、沉淀平衡状态时二氧化碳分压 (pCO2) 的变化对文石溶解、沉淀速率及其动力学方程的影响。每组实验中通入混合气体的pCO2分别为2 300×10-6和320×10-6两种,并保持通入气体的pCO2恒定,通过实验得到了这两种pCO2环境下,文石的溶解和沉淀速率及动力学方程。研究结果表明:(1) 文石在近溶解、沉淀平衡附近,溶解速率相对于Ac(碳酸盐碱度值)或Ω(饱和度)的变化速率要大于沉淀速率相对于Ac或Ω的变化速率;(2) Ac相同时,pCO2越高,溶解速率越高,沉淀速率越低;Ω相同时,亦是如此;(3) 溶解实验中,在Ω > 0.8的区间内,反应液和混合气体之间pCO2平衡,反应级数n介于9 ~ 10之间;沉淀实验中,文石的反应级数介于2.0 ~ 3.2之间。
海水;文石;溶解;沉淀;反应级数
文石是现代碳酸盐沉积物中主要矿物之一,主要分布于温暖地区的浅海灰泥沉积物及碳酸盐颗粒(如鲕粒、球粒及团块等)之中,部分出现在海滩岩、生物礁及浅海碳酸盐颗粒沉积物的胶结物中。文石也是六射珊瑚和某些软体动物介壳的典型矿物成分。由于文石质的生物骨架以及文石质的鲕粒和晶体比方解石易溶,因此成岩早期的溶解作用常具有选择性的特点。因此,模拟文石的溶解、沉淀过程对于成岩作用的定量研究以及理解碳酸盐岩溶和储层的发育有重要意义。此外,大气二氧化碳分压 (pCO2) 的明显变化[1]引起表层海水的酸化,海水pH值的降低已对海洋碳酸盐系统产生了影响[2]。研究近溶解、沉淀平衡状态pCO2的变化对文石溶解、沉淀速率及其动力学方程的影响,能加深对现今海洋碳酸盐系统的理解。
在过去几十年文石的溶解和沉淀速率及反应动力学受到了大量的关注[3-15]。研究中主要存在两个问题:(1) 溶解、沉淀平衡附近缺少可靠的数据。在靠近溶解、沉淀平衡处,由于较小的pH值误差、微小的温度或 pCO2的变化都将会对饱和度 (Ω) 的计算产生较大的影响,故而一直缺乏可靠的数据。(2) 不同的学者利用实测或模拟实验得到的反应级数n有很大差异。为了进一步认识pCO2变化对文石溶解、沉淀速率及反应级数的影响,获得可靠数据,探讨反应级数n产生差异的原因,本文利用free-drift开放反应系统进行了相关的实验。
1 材料与方法
1.1 实验试剂及仪器
a) 人工海水:人工海水的配制参照文献[16],不含PO43-离子,以排除其对反应过程的干扰[17],盐度为35;实验前未加入任何形式的碳酸盐或重碳酸盐,因此反应液初始饱和度 (Ω0) 很低,接近0;实验前,用紫外可见分光光度计(精度20 nmol/L)检测过滤的人工海水中的磷,低于检测限。
b) 超纯级文石(Sigma®):表面积测定采用Kr-BET[18]方法,S = 0.76 m2/g,“S”为每克文石的表面积。
c) CO2/N2混和气体、洗气瓶、聚四氟乙烯材质的分液漏斗状反应器 (Nalgene®,容积约660 mL)、恒温槽(Thermo®Haake,精度 ± 0.1 ℃)、温度计(精度 ± 0.1 ℃)、去离子水、过滤器、滤膜(0.45 µm)、电子天平(精度0.000 1 g)、pH电极(Orion, 8102BNUWP,精度0.001)、pH计(Mettler®235,精度0.1V)、电位滴定仪(万通798MPT Titrino)。
d) pH值测定标准采用三羟甲基氨基甲烷(Tris)和2-氨基-2-甲基-1-丙醇(AMP),Tris和AMP的配制及pH值的测定方法参照DOE(1994)[19],pH值的计算以Tris作为标准。
1.2 实验操作过程
首先,过滤人工海水约0.6 kg,准确称量后加入反应器中(图1)。接着,称取一定量的NaHCO3粉末(精确到0.000 1 g), 加入反应器中,以调节反应液Ω0。然后,从反应器底部通入pCO2恒定的CO2/N2的混合气体。混和气体的有效成分为CO2,在通入反应器前经过洗气瓶中去离子水的湿润。反应器放置在恒温槽中,保持在25 ℃,恒温槽控温精度为 ± 0.2 ℃。
为了最大程度减少反应液的蒸发,在反应器的上口部,用塑料薄板遮挡。反应器的口部留有插入温度计、电极、取液管等大小合适的孔。温度计的精度为0.1 ℃,可以实时监测反应温度。插入取液管是为了避免取样时打开反应器口部,以最大限度的维持反应液反应环境(温度、pCO2)的恒定。
当溶液和混和气体间达到 pCO2平衡(∆pH < 0.002 单位/h)时,加入文石或文石晶体粉末(精确到0.000 1 g),同时,开始计时,反应时间请见表1。每千克人工海水中加入晶体粉末的量为2.5g。从反应器底部通入的混和气体有两个作用:(1) 保持反应液具有恒定的 pCO2(对于溶解实验,后期才能真正达到 pCO2平衡);(2) 搅拌作用。通入反应液的混合气体压力稍高于反应液压力,以使文石或文石晶体粉末和反应液充分混和。在本实验中假定反应液与文石晶体粉末混和均匀,每次采样后,晶体和溶液的质量比不发生改变。
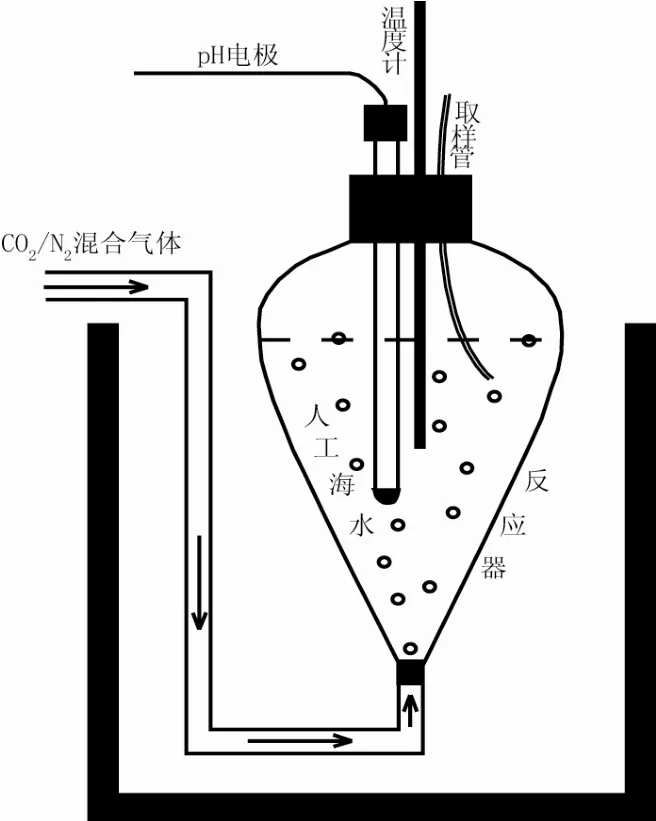
图1 溶解及沉淀反应装置Fig. 1 Dissolution and precipitation reaction equipments
1.3 取样过程
取样过程分为两个步骤:(1) 反应液pH值测定。测定方法参照DOE (1994)[19],以Tris作为标准。(2)取样:在测定反应液的pH值后,从反应器口部的取样管用注射器抽取25 ~ 30 mL样品。取样过程中要避免注射器吸入气泡,因过滤反应液时需要加压,而气泡中存在的CO2会在压力增大时加速文石或文石晶体的溶解,增加样品的海水总碱度 (At) 值,引入误差。随着溶解或沉淀过程的进行,顺序取样。
取样前先取3 ~ 5 mL反应液润洗注射器,然后将这部分反应液过滤,用于润洗样品瓶,以尽量降低器材对样品的影响。然后将抽取的样品过滤到经过润洗的样品瓶中,密封保存在冰箱中,用于以后At和 [Ca2+]的测定。
1.4 样品分析测试
利用电位滴定仪(万通 798MPT Titrino)测定样品 At以及反应液的初始钙离子浓度 ([Ca2+]0)[20]。At测定单个样品用量约25 mL,相对标准偏差低于0.10%,即± 2 µmol/kg;[Ca2+]测定单个样品用量1 mL,相对标准偏差低于0.25%,即± 26 µmol/kg,有时可达到0.10%,即± 10 µmol/kg。
2 数据的处理
2.1 碳酸体系的参数的计算
研究海洋碳酸盐体系时要涉及到四个参数:pH值,At,溶解无机碳的浓度(DIC) 和pCO2,利用其中两个参数可以计算另外两个参数。本研究测定的参数有pH值、At以及反应液的[Ca2+]0。At的定义参照
Dickson(1981)[21],碳酸盐碱度值(Ac)由At值和pH值计算得出,反应过程中的钙离子浓度( [Ca2+]t)由Ac和[Ca2+]0依据式(1)计算得出。

2.2 Ac随时间变化的曲线拟合
本文采用Gehlen等(2005)[22]运用的经验拟合方程:
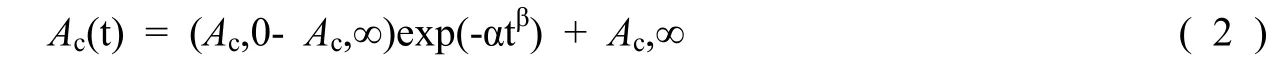
对文石在溶解、沉淀过程中Ac随时间的变化进行曲线拟合,当t = 0时,Ac,t = Ac,0; 当t→∞时,Ac,t= Ac,∞。这里α和β为自由常数,它们由最小化方差和得到。Ac相对于时间变化的曲线的斜率代表溶解或沉淀速率。因此文石的溶解速率及动力学方程可以表示为:
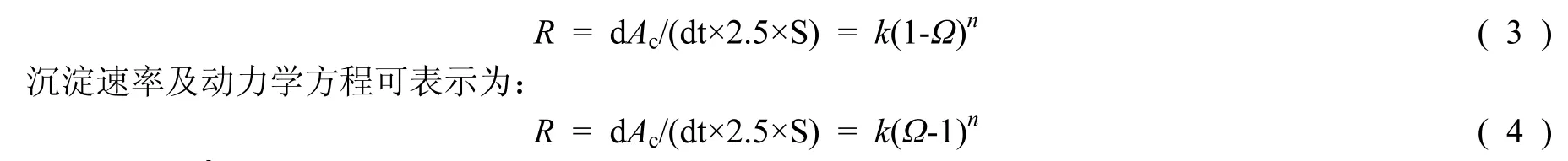
式中:S =0.76 m2/g,为发生反应的每克文石的表面积。文石晶体每平方米每年的质量溶解或沉淀速率Ry可表示为:

溶解实验中Ry为正值,沉淀实验中Ry为负值,其单位为g/(m2·a),60×24×365代表每年的分钟数,除以1 000转换为Ac摩尔数,除以2转换为碳酸盐摩尔数,乘以100转化为质量,以g为单位。可以得到:

式中:ky为文石的溶解或沉淀速率常数 (g/(m2·a)),n为反应级数。
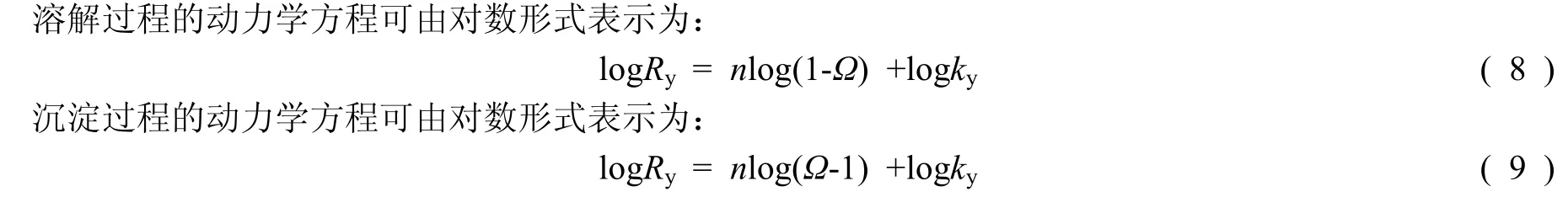
因需对 Ac随时间 t的变化进行曲线拟合,故样品点越多拟合效果越好。但由于受到反应容器体积(~ 660 mL)以及每次At检测用量(~30 mL)的限制,样品数保持在9 ~ 13个左右。样品的取样时间间隔依据经验设定,其标准为相邻两个样品间At变化值尽量保持一致。溶解及沉淀实验刚开始时,At变化快,取样间隔短,随着反应向溶解或沉淀平衡的接近,At变化也越来越慢,取样时间间隔随之增长。
3 结果与分析
3.1 溶解及沉淀实验
在恒温(25 ℃)、恒压(1×105Pa)条件下进行的文石的溶解、沉淀实验共四组,见表1。溶解实验中,为了避免开始阶段pCO2的大幅下降,准确测定pCO2平衡时文石的Ry,在实验前加入NaHCO3,把人工海水Ω0提高至0.6左右,但反应液pCO2降低依然存在,但下降幅度大大减小。表1中“最低pCO2”表示反应液 pCO2下降到的最低值。“Ω0”、“Ωe”分别是每组实验的初始饱和度和反应结束时的饱和度。“反应时间”代表每组实验所经历的时间。由于文石的沉淀是一个排气过程,实验中能很好地维持pCO2恒定。

表1 在25℃,pCO2平衡时文石溶解及沉淀实验数据表Tab. 1 Dissolution and precipitation data of aragonite with constant pCO2 at 25℃
3.2 溶解及沉淀速率
采用Gehlen等(2005)[22]运用的经验拟合方程 (2) 对文石在溶解、沉淀过程中Ac随时间的变化进行曲线拟合,拟合曲线的斜率代表溶解或沉淀速率。然后利用式 (5) 计算出文石晶体每平方米每年的质量溶解或沉淀速率Ry,溶解实验中Ry为正值,沉淀实验中Ry为负值,其单位为g/(m2·a)。
在压力和温度条件相同时研究pCO2对Ry的影响,需从Ac和Ω两方面加于考虑。反应液pCO2不同,文石的溶解度不同。因此,Ω相同时,不同的pCO2对应不同的Ac;同样,Ac相同时,不同的pCO2也对应不同的Ω。每组实验通入混合气体的pCO2恒定,但溶解实验的初始阶段,反应液的pCO2会发生轻微下降,在计算Ry时可忽略,而沉淀过程中反应液pCO2始终保持恒定。由图2可知:(1) 文石在近溶解、沉淀平衡附近,溶解速率相对于Ac的变化速率要大于沉淀速率相对于Ac的变化速率;(2) Ac相同时,pCO2越高,溶解速率越高,沉淀速率越低;(3) 在相同的pCO2环境下,溶解实验和沉淀实验在横轴的交点代表此环境下溶解度所对应的Ac值。
图3是近溶解、沉淀平衡处,Ry随Ω的变化。由图3可知:(1) 文石在近溶解、沉淀平衡附近,溶解速率相对于Ω的变化速率要大于沉淀速率相对于Ω的变化速率; (2) 在相同Ω条件下,反应液pCO2越高,文石溶解速率越快,沉淀速率越慢。
3.3 溶解及沉淀动力学
为了进一步说明不同pCO2条件下,Ry和Ω之间的关系,在溶解实验中做Ry和 (1-Ω) 之间对数关系图,在沉淀实验做Ry和 (Ω-1) 之间对数关系图(图4)。因溶解实验的初始阶段,存在反应液的pCO2降低(反应液最低pCO2请见表1),所以可把溶解实验分为两段,即反应液pCO2降低阶段(阶段I)和反应液pCO2恒定阶段(阶段II)。沉淀实验及溶解实验两阶段的动力学方程请见表1。
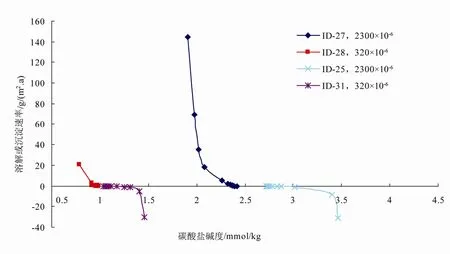
图2 在文石近溶解、沉淀平衡处,Ac对Ry的影响Fig. 2 Influence of Ac on Ry for aragonite near dissolution and precipitation equilibrium
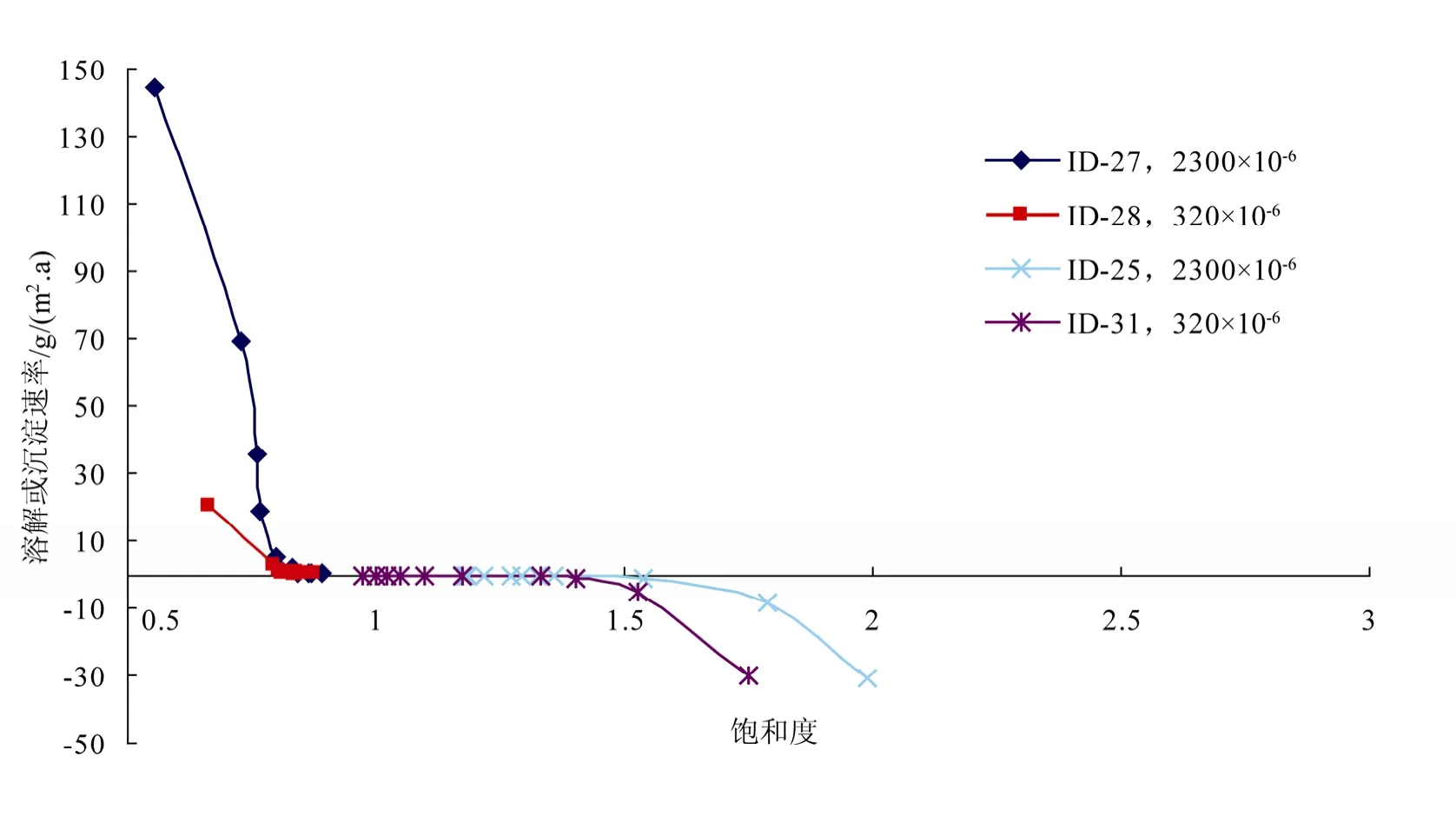
图3 在文石近溶解、沉淀平衡处,pCO2对Ry的影响Fig. 3 Influence of pCO2 on Ry for aragonite near dissolution and precipitation equilibrium
对应图4,从表1可知,溶解实验在pCO2欠平衡阶段(阶段I),反应级数n小于5,而pCO2平衡阶段(阶段II),Ω > 0.8,反应级数n介于9 ~ 10之间。在阶段I,ID-27和ID-28的反应级数相差较大,其原因是由Ω0不同造成的。因Ω0ID-27< Ω0ID-28, 使得ID-21在溶解初始阶段反应液的pCO2下降更明显,使得pCO2欠平衡阶段的反应级数更小。沉淀实验中,文石的反应级数介于2.0 ~ 3.2之间。
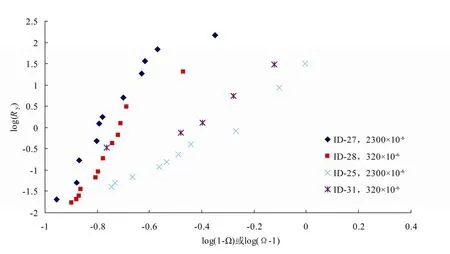
图4 pCO2恒定时,近平衡条件下文石的Ω和Ry之间的对数关系Fig. 4 Logarithmic relationship between Ω and Ry for aragonite near dissolution equilibrium with constant pCO2
4 结 论
在文石的溶解、沉淀实验中,通过精确控制反应过程的温度、降低pH值误差、提高海水总碱度值测定的精度,获得了溶解、沉淀平衡附近可靠的数据。在溶解实验中Ω > 0.8的区间内,反应液和混合气体之间pCO2平衡(阶段II),反应级数n介于9 ~ 10之间,远大于Keir[11]所得到的反应级数4.2。这很可能是由于其未充分考虑反应液和混合气体之间pCO2欠平衡阶段(阶段I),把阶段I和阶段II(表1)笼统的作为一个整体,同时由于其实验的取样点多位于阶段 I,即溶解平衡附近数据点较少,从而导致其所得到的n值偏小。此外,Ω较低时与Ω较高时的溶解速率控制机制不同,前者为扩散作用控制机制 (diffusion controlled reaction),后者为固体表面反应控制机制 (surface controlled reaction)[23],不同的反应机制导致这两个阶段n值存在较大的差异。在沉淀实验中,n值介于2.0 ~ 3.2之间,这对研究无机文石矿物的形成条件以及地史时期“文石海”的形成具有一定的借鉴意义。
[1] Körtzinger A, Mintrop L, Wallace D W R. The international at-sea intercomparison of fCO2systems during the R/V Meteor Cruise 36/1 in the North Atlantic Ocean [J]. Marine Chemistry, 2000, 72(2-4): 171-192.
[2] Feely R A, Sabine C L, Lee K, et al. Impact of anthropogenic CO2on the CaCO3system in the oceans [J]. Science,2004. 305:362-366.
[3] Berner R A, Wilde P. Dissolution kinetics of calcium carbonate in sea wAter:I.Saturation state parameters for kinetic calculations [J]. American Journal of Science, 1972, 272: 826-839.
[4] Morse J W, Berner R A. Dissolution kinetics of calcium carbonate in seawater: II. A kinetic origin for lysocline [J]. American Journal of Science, 1972,272: 840-851.
[5] Berner R A, Morse J W. Dissolution kinetics of calcium carbonate in sea water: IV. Theory of calcite dissolution [J]. American Journal of Science, 1974,274: 108-134.
[6] Morse J W. Dissolution kinetics of calcium carbonate in seawater: V. Effects of natural inhibitors and the position of the chemical lysocline [J].American Journal of Science, 1974, 274: 638-647.
[7] Morse J W. Dissolution kinetics of calcium carbonate in seawater: IV. The near-equilibrium dissolution kinetics of calcium carbonAte-rich deep sea sediments [J]. American Journal of Science, 1978, 278: 344-353.
[8] Morse J W, Mucci A, Millero F J. The Solubility of calcite and aragonite in seawater of 35 per thousand aalinity at 25℃ and atmospheric pressure [J].Geochimica et Cosmochimica Acta, 1980, 44(1): 85-94.
[9] Mucci A. The solubility of calcite and aragonite in seawater at various salinities, temperatures, and one atmosphere total pressure [J]. American Journal of Science, 1983, 283: 780-799.
[10] Walter L M, Morse J W. Reactive surface area of skeletal carbonates during dissolution; effect of grain size [J]. Journal of Sedimentary Research,1984, 54(4): 1 081-1 090.
[11] Keir R S. The dissolution kinetics of biogenic calcium carbonates in seawater [J]. Geochimica et Cosmochimica Acta, 1980, 44: 241-252.
[12] Kralj D, Vdovi N. The influence of some naturally occurring minerals on the precipitation of calcium carbonate polymorphs [J]. Water Research, 2000,34(1): 179-184.
[13] Morse J W. The kinetics of calcium carbonate dissolution and precipitation [J]. In: R.J. Reeder, Editor, Carbonates Mineralogy and Chemistry, Reviews in Mineralogy 11. Mineralogical Society of America, 1983: 227-264.
[14] Wei Hao, Shen Qiang, Zhao Ying, et al. Influence of polyvinylpyrrolidone on the precipitation of calcium carbonate and on the transformation of vaterite to calcite [J]. Journal of Crystal Growth, 2003, 250(3-4): 516-524.
[15] Wray J L, Daniels F. Precipitation of calcite and aragonite [J]. Journal of the American Chemical Society, 1957, 79: 2 031-2 034.
[16] Kester D P, Duedall I W, Connors D N, et al. Preparation of artificial seawater [J]. Limnology and Oceanography, 1967, 12(1): 176-179.
[17] Mucci A. Growth kinetics and composition of magnesian calcite overgrowths precipitated from seawater—Quantitative influence of ortho-phosphate ions [J]. Geochimica et Cosmochimica Acta, 1986, 50: 2 255–2 265.
[18] Gregg S J, Sing K S W. Adsorption, surface area, and porosity [M]. Academic Press(London), 1982: 303.
[19] DOE. Handbook of methods for the analysis of the various parameters of the carbon dioxide system in seawater (version 2)[Z].In: Dickson, A.G., Goyet,C. (Eds.),ORNL/CDIAC-74. Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, US Department of Energy, Oak Ridge,Tennessee.1994.
[20] Tao Xiaowan, Pu Xiaoqiang, Ni Yunyan, et al. Determination of total alkalinity and calcium concentration of seawater rapidly and automaticly with small-amount samples[C]. Special Track: Environmental Pollution and Public Health (EPPH 2009) within the 3rd International Conference on Bioinformatics and Biomedical Engineering, iCBBE, 2009 (accepted).
[21] Dickson A G. An exact definition of total alkalinity and a procedure for the estimation of alkalinity and total inorganic carbon from titration data [J].Deep-Sea Research, 1981, 28(A): 609-623.
[22] Gehlen M, Bassinot F C, Chou L,et al. Reassessing the dissolution of marine carbonates: II. Reaction kinetics [J]. Deep-Sea Research I, 2005, 52:1 461–1 476.
[23] Morse J M, Arvidson R S. The dissolution kinetics of major sedimentary carbonate minerals [J]. Earth-Science Reviews, 2002, 58: 51-84.
Research on dissolution and precipitation rates and kinetics of aragonite in artificial seawater
TAO Xiao-wan1, LUO Chun-shu2, CHEN Li-xin2, WANG Xian-chao3
(1. Research Institute of Petroleum Exploration & Development, PetroChina, Beijing 100083, China;2. Research Institute of Petroleum Exploration & Development, Tarim Oilfield Company, PetroChina, Korla 510640, China;3. Shandong Zhongmei Parent Corporation of Coal Geophysical Prospecting & Surveying, Tai’an 271000, China)
The influence of variations of carbon dioxide partial pressure (pCO2) in seawater on the precipitation rate and kinetics of calcite and aragonite was studied using free-drift open reaction system which was kept at constant pressure (1atm) and temperature (25.0±0.2℃). In the experiments, two kinds of mixture gas with pCO2≈2300×10-6and pCO2≈320×10-6were used respectively. In every experiment, the pCO2of mixture gas was kept constant. And the dissolution and precipitation rates and reaction equations were achieved under different pCO2. The results show that: (1)Near dissolution and precipitation equilibrium of aragonite, the variation rate of dissolution rate in relative of carbonate alkalinity (Ac) or saturation state (Ω) is larger than that of precipitation rate in relative of carbonate alkalinity(Ac) or saturation state(Ω). (2) The higher the pCO2, the higher the dissolution rate and lower precipitation rate of aragonite with the same Acor Ω. (3) In dissolution experiments, the equilibrium of pCO2between reaction solution and mixture gas is achieved when Ω>0.8, and the reaction order is between 9 and 10 for aragonite. And in precipitation experiments,the reaction order is between 2.0 to 3.2 for aragonite.
seawater; aragonite; dissolution; precipitation; reaction order
P736.4+2
A
1001-6932(2010)02-0143-07
2009-05-06;
2009-08-20
国家自然科学基金(40376038)资助
陶小晚 (1982-),男,博士研究生,研究方向为地球化学方面的研究工作,电子邮箱:taoxiaowan000112@163.com
