Delayed hepatocarcinogenesis through antiangiogenic intervention in the nuclear factor-kappa B activation pathway in rats
2010-12-14ZhiZhenDongDengFuYaoWeiWuMinYaoHongBoYuJunJunShenLiWeiQiuNingHuaYaoWenLiSaiandJunLingYang
Zhi-Zhen Dong, Deng-Fu Yao, Wei Wu, Min Yao, Hong-Bo Yu, Jun-Jun Shen,Li-Wei Qiu, Ning-Hua Yao, Wen-Li Sai and Jun-Ling Yang
Nantong, China
Delayed hepatocarcinogenesis through antiangiogenic intervention in the nuclear factor-kappa B activation pathway in rats
Zhi-Zhen Dong, Deng-Fu Yao, Wei Wu, Min Yao, Hong-Bo Yu, Jun-Jun Shen,Li-Wei Qiu, Ning-Hua Yao, Wen-Li Sai and Jun-Ling Yang
Nantong, China
(Hepatobiliary Pancreat Dis Int 2010; 9: 169-174)
hepatocellular carcinoma;nuclear factor-kappa B;vascular endothelial growth factor;intervention;dynamic expression
Introduction
Carcinogenesis of hepatocellular carcinoma(HCC) is a multi-factor, multi-step, and complex process,[1,2]and many genes such as protooncogenes, tumor suppressor genes, apoptosis genes,and growth factor genes have been implicated; apoptosis genes may play an important role in the process of HCC.[3]Nuclear factor-kappa B (NF-κB) is a crucial regulator of innate immune and in fl ammatory responses,cell proliferation, and apoptosis through activation of responsive genes.[4]In resting cells, NF-κB is present as a latent, inactive complex retained in the cytoplasm through association with inhibitory proteins (IκB).[5]It has a pleiotropic effect, controlling the expression of numerous genes with multi-directional regulation; it regulates the transcription and expression of diverse genes encoding cytokines, growth factors, adhesion molecules, and pro- and anti-apoptotic proteins.[6]Abnormally activated NF-κB modulates the transcription and expression of genes, and plays an important role in the development of HCC.[7,8]
NF-κB binding to IκB segregates NF-κB into the cytoplasm and forms a trimer, preventing NF-κB from directly receiving nuclear transposition signal stimulation by interaction with proteins. IκBα,being both target gene and regulatory factor of NF-κB, directly inhibits the DNA binding activity of NF-κB, and is phosphorylated and degraded under the in fl uence of cytokines, ultraviolets.[9,10]Moreover, in response to many extracellular stimuli such as infection with hepatitis B virus (HBV) or hepatitis C virus,IκB proteins are phosphorylated by the IκB kinase complex.[11,12]The released NF-κB rapidly enters the nucleus where it performs its function.[4,13]However,the relationship between abnormal NF-κB activation and malignant transformation of hepatocytes remains to be explored.[5,14,15]In this study, we investigated the expression of NF-κB and vascular endothelial growth factor (VEGF) at different stages of HCC formation,and explored the in fl uences on HCC through the antiangiogenic inhibition of NF-κB activation.
MethodsHepatoma model and drug intervention
Eighty-four male Sprague-Dawley rats, weighing 120-160 g, provided by the Animal Center of Medical Experiments, Nantong University, China, were randomly divided into three groups: control (n=12, 6 rats×2),2- fl uorenylacetamide (2-FAA; Sigma, USA) (n=36, 6 rats×6), and 2-FAA plus thalidomide (n=36, 6 rats×6).The rats in the control and 2-FAA groups were treated according to a previously reported method.[16]The rats in the 2-FAA plus thalidomide group were given food containing 2-FAA (0.05%), and simultaneously 100 mg/kg of thalidomide (No. 0710081, Changzhou Pharmaceutical Co., China) dissolved in olive oil administered intragastrically from the next day. Six rats in the 2-FAA group or in the 2-FAA plus thalidomide group and two rats in the control group were sacri fi ced every two weeks by anesthesia with diethyl ether. Blood was drawn from the heart. Parts of liver specimens were used for pathological and immunohistochemical analysis or extraction of total RNA, and the rest were kept at -80 ℃ until use. All procedures were conducted in accordance with the Guidelines for Experimental Animals approved by the Animal Care and Use Committee of Nantong University, China.
Immunohistochemistry
Liver tissues were formalin- fi xed and paraf fi nembedded for histological examination, or subjected to the streptavidin-peroxidase (S-P) method according to standard procedures. PBS was used to substitute for the primary antibody as a negative control. NF-κB and VEGF stainings were evaluated semi-quantitatively on the basis of the percentage of positive cells.[17]
Extraction of liver proteins
Fifty milligrams of rat liver tissue was homogenized in 1 ml of PBS for 15 minutes at 4 ℃, stored at 4 ℃overnight, and centrifuged at 12 000 g for 15 minutes.The supernatant was transferred to a new 1.5 ml Eppendorf tube for analysis.
Detection of VEGF in liver tissues
The levels of VEGF in HCC tissues were determined at 492 nm by quantitative colorimetric sandwich enzyme linked immunosorbent assay (ELISA; R&D systems, UK)in accordance with the manufacturer's instructions.[18]Concentrations were calculated using a standard curve generated with speci fi c standards provided by the manufacturer.
Western blotting
The proteins in the supernatants were subjected to Western blotting as described previously.[18]Brie fl y, for each sample, 50 μg of protein was separated using 10%SDS-polyacrylamide gel electrophoresis. The proteins were then transferred onto a PVDF membrane and incubated with the primary antibody (rabbit anti-rat)in Tris-Buffered Saline Tween (TBST)-20 for 2 hours at room temperature (BD Biosciences Pharmingen, CA),along with beta-actin (New England Bio Labs, MA).The membranes were then incubated with a secondary antibody (goat anti-rabbit; New England Bio Labs)in TBST also for 1 hour at room temperature. The immuno-complexes were visualized using an ECL kit(Amersham Pharmacia Biotech, UK) and images were developed with the Molecular Imager® Gel DocTMXR System (Bio-Rad laboratories, Inc., USA).
Extraction of nuclear proteins and detection of NF-κB
Fifty milligrams of fresh tissue was homogenized with a Polytron homogenizer according to the instructions with a nuclear and cytoplasmic protein extraction kit (Beyotime Co., China). The nuclear proteins were quanti fi ed spectrophotometrically using a bicinchoninic acid assay kit (BCA, Beyotime Co., China). NF-κB levels were detected using a rat NF-κB ELISA kit (Shanghai Xitang Biotechnology Co., China) according to the manufacturer's instructions. Concentrated detergent and specimens were diluted 20-fold. Finally, 100 μl termination buffer was added, mixed lightly for 30 seconds, and the absorbance read at 490 nm. The concentration of NF-κB was calculated according to the standard.
NF-κB cDNA synthesis and ampli fi cation
Fifty milligrams of liver tissue was homogenized with a Polytron homogenizer after the addition of 1.0 ml of Trizol reagent (Promega), and then processed according to the manufacturer's instructions. Two sets of primers were designed according to the NF-κB sequence (NM_199267) and synthesized (Shanghai Institute of Cell Biology, China). The sequences of the two external primer pairs used for the initial PCR ampli fi cation were NF-κB-P1: 5'-ACC AAA GAC CCA CCT CAC C-3' (nt 293-311), and NF-κB-P2: 5'-CGC ATT CAA GTC ATA GTC CC-3' (nt 510-529). The sequences of the two internal primer pairs used for the second PCR ampli fi cation were NF-κB-P3: 5'-GGG ATG GCT TCT ATG AGG CT-3' (nt 348-367) and NF-κB-P4:5'-TGA AAG GGG TTA TTG TTG GTC-3' (nt 466-486).
For synthesis of cDNA, 2 μg of total RNA was denatured in the presence of random hexamer (100 pmol/L, Promega) and reverse-transcriptase (GIBCO,BRL) at 23 ℃ for 10 minutes, 42 ℃ for 60 minutes,and 95 ℃ for 10 minutes, then on ice for 5 minutes,and stored at -20 ℃ for PCR ampli fi cation. The PCR ampli fi cation consisted of initial denaturation at 94 ℃ for 5 minutes, followed by 94 ℃ for 25 seconds,55 ℃for 30 seconds, and 72 ℃ for 90 seconds for 30 cycles. The fi nal product of nested PCR was 139bp. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH)gene was used for control. The primer sequences for GAPDH were GAPDH-1 (sense), 5'-ACC ACA GTC CAT GCC ATC AC-3' (nt 601-620) and GAPDH-2 (antisense),5'-TCC ACC ACC CTG TTG CTG TA-3' (nt 1033-1052),and the PCR product was 452bp. All PCR products were electrophoresed on 2% agarose gels and stained with ethidium bromide. The fragment sizes were evaluated using PCR markers (Promega) as molecular weight standards.
Statistical analysis
Data were expressed as mean±SD. Statistical analysis was carried out with SPSS version 11.0. Differences between groups were assessed by Student's t test or the Chi-square test. P<0.05 was regarded as statistically signi fi cant.
ResultsPathological alterations and occurrence of HCC
The pathological alterations of rat liver in the 2-FAA group were the same as in previous reports. The comparison of rat liver morphological changes between the 2-FAA plus thalidomide group and the 2-FAA group is shown in Table 1. At the early stage, the structure of liver lobules was intact, with only a few hepatocytes showing punctiform denaturation and necrosis(50%, 18/36 vs. 86.1%, 31/36). At the middle stage,the structure of liver lobules was still basically intact,without large fl ake-like necrosis, and some hepatocytes proliferated mildly (25%, 9/36 vs. 13.9%, 5/36). At the fi nal stage, the structure of liver lobules was still present in most hepatocytes, nodular hyperplasia was observed in some areas, and no cancer was found in the 2-FAA plus thalidomide group (25%, 9/36 vs. 0%, 0/36), where the structure of liver lobules was damaged in only a few rats, whose hepatocytes showed multi-nodular hyperplasia. And only one rat generated atypical hyperplasia. A signi fi cant difference was found between the 2-FAA group and the 2-FAA plus thalidomine group (P<0.01), suggesting that the antiangiogenic drug delayed the occurrence of HCC, as con fi rmed by pathological examination.
Expression of NF-κB and VEGF in hepatocarcinogenesis
The immunohistochemical analysis of NF-κB or VEGF in rat hepatocarcinogenesis is summarized in Table 2. The positive NF-κB staining in the rat livers was mainly located in the cytoplasm as brown granules, and particles localized in the nucleus. However, the positive brown granules staining for VEGF were mainly located in the cytoplasm. The incidence of NF-κB and VEGF expression was 88.9% and 83.3% in the denatured group and both were positive in all members of the precancerous and cancerous groups. The incidence and intensity of hepatic NF-κB or VEGF expression at anystage in the experimental group was higher than that in the control group (P<0.001). The ampli fi cation analysis of NF-κB mRNA (Fig. 1A), the speci fi c concentration of liver NF-κB (Fig. 1B), and the Western blotting of VEGF(Fig. 1C) con fi rmed that they were clearly increased in the development of hepatomas, but not in the controls,showing signi fi cant differences between the cancerous or precancerous groups and the control group.
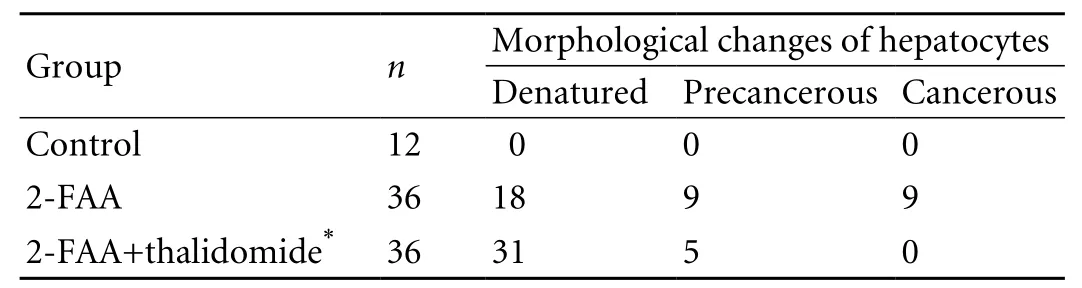
Table 1. Histological changes (HE staining) of rat hepatocytes in the 2-FAA and 2-FAA plus thalidomide groups
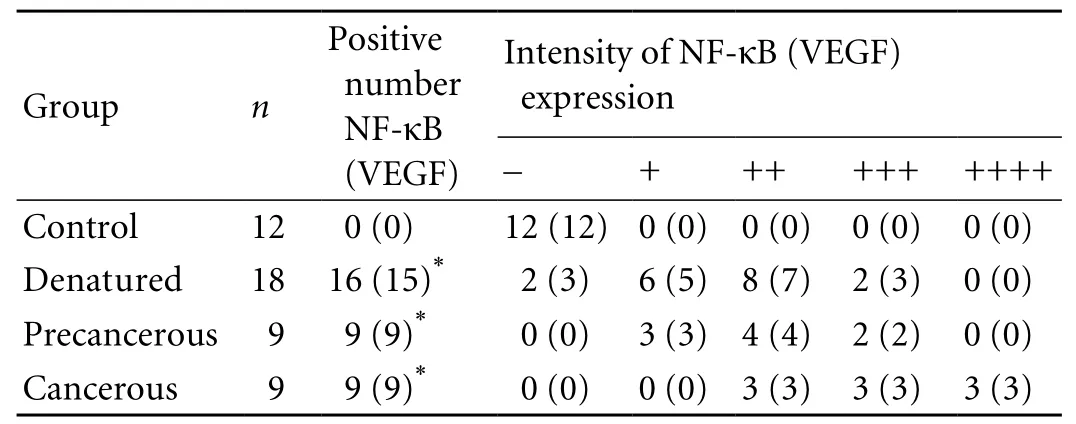
Table 2. Dynamic expression of NF-κB and VEGF in rat hepatocarcinogenesis
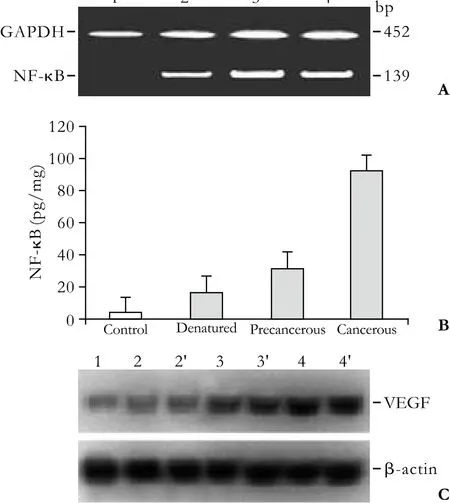
Fig. 1. Alterations of NF-κB and VEGF expression at different stages of rat hepatoma development. A: NF-κB mRNA was synthesized to NF-κB cDNA with random hexamer and Moloney murine leukemia virus reverse-transcriptase, and detected with different primer pairs by nested PCR (139bp). The amplified fragments of the NF-κB gene were distinct at the different stages of hepatoma development. NF-κB mRNA in rat liver: lane 1,control; lane 2, denatured; lane 3, precancerous; lane 4, cancerous.The GAPDH gene (452bp) was used for control. B: Quantitative analysis of hepatic NF-κB concentration at the different stages of hepatoma development. C: Western blotting of hepatic VEGF protein at the different stages of hepatoma formation. Lane 1,control; lane 2, 2', denatured; lane 3, 3', precancerous; lane 4, 4',cancerous.
Inhibition of NF-κB and VEGF expression by antiangiogenic drug
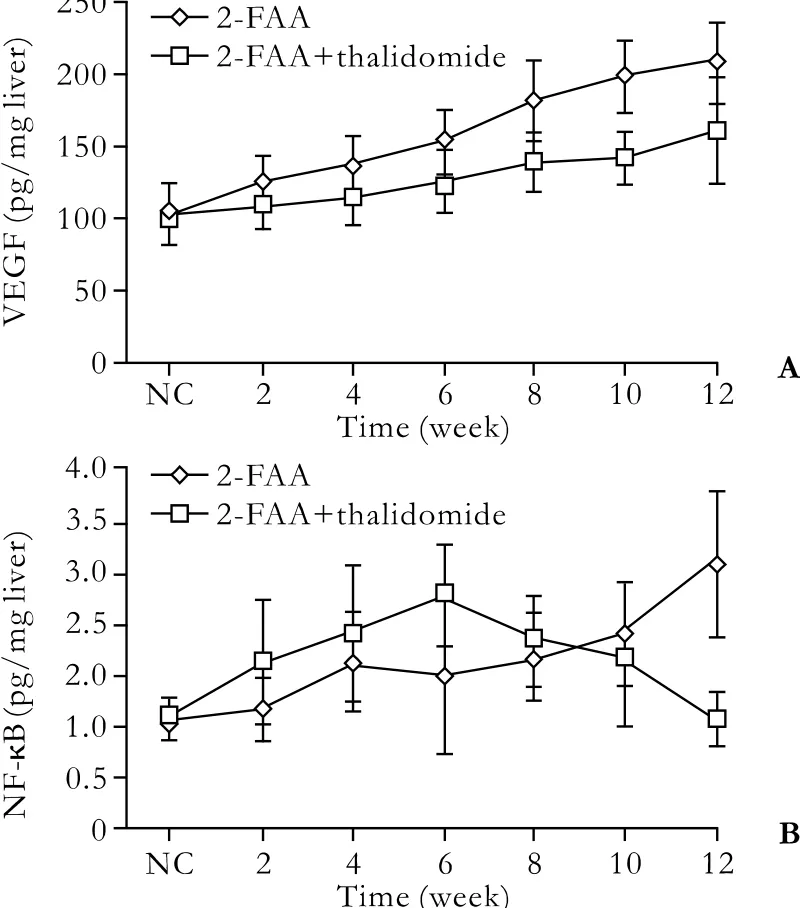
Fig. 2. Dynamic alterations of hepatic VEGF (A) and NF-κB (B)expression during the course of antiangiogenic therapy. NC, the normal control.
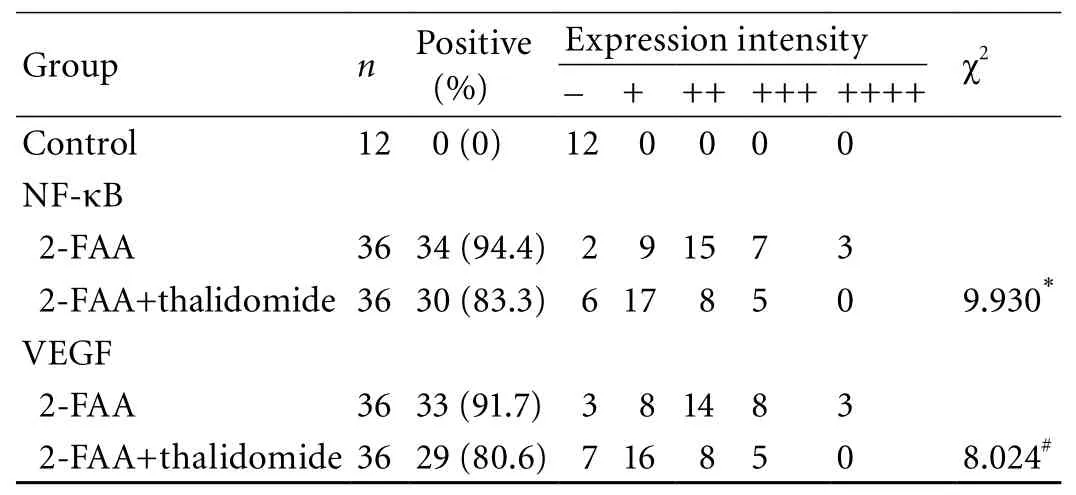
Table 3. Comparative analysis of NF-κB and VEGF expression intensity in the 2-FAA plus thalidomide and 2-FAA groups
The dynamic expression of hepatic NF-κB and VEGF between the 2-FAA plus thalidomide group and the 2-FAA group are shown in Fig. 2. The expression of hepatic VEGF in the 2-FAA plus thalidomide group was inhibited during the development of hepatoma. The NF-κB expression in the 2-FAA plus thalidomide group rose at fi rst gradually, when it was higher than that in the 2-FAA group in the same period, peaked at week 6,and then declined gradually to normal at week 12. The differences of VEGF and NF-κB between the 2-FAA plus thalidomide group and the 2-FAA group are shown in Table 3. The expression intensity of NF-κB in the 2-FAA plus thalidomide group was lower than that in the 2-FAA group (χ2=9.930,<0.001), and the expression intensity of VEGF in the 2-FAA plus thalidomide group was signi fi cantly lower than that in the 2-FAA group(χ2=8.024, P<0.001), indicating that the antiangiogenic drug down-regulated NF-κB expression and decreased angiogenesis.
Discussion
HCC is one of the most common malignancies worldwide, but treatment outcomes have remained generally poor.[19,20]In China, about 90% of HCC cases are associated with HBV infection.[21,22]The active form of NF-κB is involved in the initiation, generation,and development of tumors, and is up-regulated in malignancies associated with in fl ammation.[1,23]NF-κB activation in in fl ammation-related liver diseases regulates the expression of in fl ammation response- or cancerrelated genes.[3,6,24]In this study, we intragastrically administered an antiangiogenic drug to intervene in NF-κB activation, and assessed its in fl uence on malignant transformation of hepatocytes in rat hepatocarcinogenesis.
The liver tissues of rats fed with 2-FAA showed vacuole-like denaturation at the early stage, subsequent appearance of dysplastic nodules at the middle stage,and fi nally progressed to tubercles of cancerous nests.NF-κB expression in the denatured, precancerous,and cancerous groups was signi fi cantly higher than in the control group. The abnormally activated NF-κB went from the cytoplasm into the nucleus to modulate gene transcription, and fi nally contributed to hepatocarcinogenesis, indicating that NF-κB activation was dynamically increased and might play an important role in HCC development.[15]
Thalidomide is a glutamic acid analogue and has been investigated in the antiangiogenic therapy of HCC.[25,26]In the 2-FAA group, the hepatocytes showed large fl ake necrosis at week 2. Then a remarkable compensatory hyperplasia occurred and the structure of liver lobules was severely damaged. Precancerous and cancerous lesions started to appear at week 8. In the thalidomide group, before week 8, only a little focal necrosis occurred, and the structure of liver lobules was basically intact. At week 10, mild and local nodular hyperplasia appeared, and the structure of liver lobules was still basically intact. And at week 12, more nodular hyperplasia and a little atypical hyperplasia formed, and the structure of the liver lobules was partly destroyed.The density of NF-κB expression in the 2-FAA plus thalidomide group was lower than that in the 2-FAA group. In the 2-FAA plus thalidomide group, NF-κB at fi rst gradually increased, and its level was higher than that in the 2-FAA group in the same period, peaked at week 6, then declined gradually, and dropped to normal at week 12, suggesting that the intervention of thalidomide in hepatocarcinogenesis effectively inhibits or delays the angiogenesis, hyperplasia, and cancer development of hepatocytes.
Thalidomide might promote NF-κB activation by mechanisms at the initial stage, and the activated NF-κB in the nucleus is greatly increased, prevent the occurrence of hepatoma by suppressing the necrosis of hepatocytes. This was veri fi ed by the signi fi cant differences in histological changes between the 2-FAA plus thalidomide and the 2-FAA groups at the initial stage. But then the activation of NF-κB was suppressed by thalidomide, so the level of NF-κB in the nucleus markedly decreased. Because of the excessive drop of NF-κB, the protection of hepatocytes against denaturation and necrosis was lost. Afterward the necrosis, proliferation,and dysplasia of rat hepatocytes started to appear.[4,13]Thalidomide signi fi cantly suppressed the malignant transformation of hepatocytes through inhibition of NF-κB activation, down-regulated VEGF expression,and decreased angiogenesis.[23,27,28]
HBV infection is involved in human HCC progression,and activates a variety of signaling pathways, including NF-κB.[29]NF-κB is involved in similar biological processes in cancers, as a critical modulator of genes that promote cell survival, in fl ammation, angiogenesis, progression, and metastasis.[30]The environment enhances cell proliferation, survival, and migration, as well as angiogenesis, thereby promoting tumor development. NF-κB plays a pivotal role in the malignant transformation of hepatocytes. Then inhibition of NF-κB activation and down-regulation of VEGF expression may be an ideal antiangiogenic therapy for HCC.[31,32]
Acknowledgements
We are grateful to Professor Gong-Sheng Shi (Department of Pathology, Af fi liated Hospital of Nantong University) for his excellent technical assistance in an immunohistochemical study and Professor Yi-Xiang Shao (Experimental Animal Center of Nantong University) for his help in the breeding of rats.
Funding: This study was supported by grants from the Project of Elitist Peak in Six Fields (No. 2006-B-063), and the Project of Medical Sciences (H200727), the Bureau of Health, Jiangsu Province, China.Ethical approval: Not needed.
Contributors: DZZ, WW, and YDF proposed and wrote the fi rst draft. All authors contributed to the design and interpretation of the study and further drafts. YDF is the guarantor.
Competing interest: No bene fi ts in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J, Avila MA. In fl ammation and liver cancer: new molecular links. Ann N Y Acad Sci 2009;1155:206-221.
2 Lupberger J, Hildt E. Hepatitis B virus-induced oncogenesis.World J Gastroenterol 2007;13:74-81.
3 Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002;31:339-346.
4 Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev 2006;210:171-186.
5 Arsura M, Cavin LG. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett 2005;229:157-169.
6 Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 2006;441:431-436.
7 Chan CF, Yau TO, Jin DY, Wong CM, Fan ST, Ng IO. Evaluation of nuclear factor-kappaB, urokinase-type plasminogen activator, and HBx and their clinicopathological signi fi cance in hepatocellular carcinoma. Clin Cancer Res 2004;10:4140-4149.
8 Zhang X, Liu S, Hu T, Liu S, He Y, Sun S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fi bronectin expression. Hepatology 2009;50:490-499.
9 Vainer GW, Pikarsky E, Ben-Neriah Y. Contradictory functions of NF-kappaB in liver physiology and cancer.Cancer Lett 2008;267:182-188.
10 O'Neil BH, Bůzková P, Farrah H, Kashatus D, Sanoff H,Goldberg RM, et al. Expression of nuclear factor-kappaB family proteins in hepatocellular carcinomas. Oncology 2007;72:97-104.
11 Guo K, Kang NX, Li Y, Sun L, Gan L, Cui FJ, et al. Regulation of HSP27 on NF-kappaB pathway activation may be involved in metastatic hepatocellular carcinoma cells apoptosis. BMC Cancer 2009;9:100.
12 Cheng JC, Chou CH, Kuo ML, Hsieh CY. Radiationenhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction pathway. Oncogene 2006;25:7009-7018.
13 Park SG, Lee T, Kang HY, Park K, Cho KH, Jung G. The in fl uence of the signal dynamics of activated form of IKK on NF-kappaB and anti-apoptotic gene expressions: a systems biology approach. FEBS Lett 2006;580:822-830.
14 Auyeung KK, Law PC, Ko JK. Astragalus saponins induce apoptosis via an ERK-independent NF-kappaB signaling pathway in the human hepatocellular HepG2 cell line. Int J Mol Med 2009;23:189-196.
15 Van Waes C. Nuclear factor-kappaB in development,prevention, and therapy of cancer. Clin Cancer Res 2007;13:1076-1082.
16 Yao DF, Jiang DR, Wu XH. Experimental study on value of carcino-embryonic gama-glutamyl transferase isoenzymes for monitoring carcinogenesis of hepatocytes. Zhonghua Gan Zang Bing Za Zhi 2000;8:30-32.
17 Yao DF, Wu XH, Zhu Y, Shi GS, Dong ZZ, Yao DB, et al.Quantitative analysis of vascular endothelial growth factor,microvascular density and their clinicopathologic features in human hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int 2005;4:220-226.
18 Wang Z, Zhou J, Fan J, Qiu SJ, Yu Y, Huang XW, et al. Effect of rapamycin alone and in combination with sorafenib in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res 2008;14:5124-5130.
19 Chung C, Park SG, Park YM, Joh JW, Jung G. Interferongamma sensitizes hepatitis B virus-expressing hepatocarcinoma cells to 5- fl uorouracil through inhibition of hepatitis B virus-mediated nuclear factor-kappaB activation. Cancer Sci 2007;98:1758-1766.
20 Sato Y, Kato J, Takimoto R, Takada K, Kawano Y, Miyanishi K, et al. Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut 2006;55:1801-1808.
21 Lee SH, Park SG, Lim SO, Jung G. The hepatitis B virus X protein up-regulates lymphotoxin alpha expression in hepatocytes. Biochim Biophys Acta 2005;1741:75-84.
22 Lee TH, Tai DI, Cheng CJ, Sun CS, Lin CY, Sheu MJ, et al. Enhanced nuclear factor-kappa B-associated Wnt-1 expression in hepatitis B- and C-related hepatocarcinogenesis:identi fi cation by functional proteomics. J Biomed Sci 2006;13:27-39.
23 Wang J, Tokoro T, Higa S, Kitajima I. Anti-in fl ammatory effect of pitavastatin on NF-kappaB activated by TNF-alpha in hepatocellular carcinoma cells. Biol Pharm Bull 2006;29:634-639.
24 Guo LL, Xiao S, Guo Y. Activation of transcription factors NF-kappaB and AP-1 and their relations with apoptosis associated-proteins in hepatocellular carcinoma. World J Gastroenterol 2005;11:3860-3865.
25 Pinter M, Wichlas M, Schmid K, Plank C, Müller C, Wrba F,et al. Thalidomide in advanced hepatocellular carcinoma as antiangiogenic treatment approach: a phase I/II trial. Eur J Gastroenterol Hepatol 2008;20:1012-1019.
26 Yau T, Chan P, Epstein R, Poon RT. Evolution of systemic therapy of advanced hepatocellular carcinoma. World J Gastroenterol 2008;14:6437-6441.
27 Bar-Yehuda S, Stemmer SM, Madi L, Castel D, Ochaion A,Cohen S, et al. The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via deregulation of the Wnt and NF-kappaB signal transduction pathways. Int J Oncol 2008;33:287-295.
28 Calvisi DF, Pascale RM, Feo F. Dissection of signal transduction pathways as a tool for the development of targeted therapies of hepatocellular carcinoma. Rev Recent Clin Trials 2007;2:217-236.
29 Qiao L, Zhang H, Yu J, Francisco R, Dent P, Ebert MP, Rocken C, et al. Constitutive activation of NF-kappaB in human hepatocellular carcinoma: evidence of a cytoprotective role.Hum Gene Ther 2006;17:280-290.
30 Karin M. NF-kappaB and cancer: mechanisms and targets.Mol Carcinog 2006;45:355-361.
31 Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ 2006;13:738-747.
32 Wu JM, Sheng H, Saxena R, Skill NJ, Bhat-Nakshatri P, Yu M, et al. NF-kappaB inhibition in human hepatocellular carcinoma and its potential as adjunct to sorafenib based therapy. Cancer Lett 2009;278:145-155.
BACKGROUND: The active form of nuclear factor-kappa B (NF-κB) is involved in the initiation, generation, and development of hepatocellular carcinoma (HCC), and is up-regulated in in fl ammation-associated malignancies. We investigated the dynamic expression of NF-κB and its in fl uences on the occurrence of HCC through antiangiogenic (thalidomide)intervention in NF-κB activation.
METHODS: Hepatoma models were induced with 2- fl uorenylacetamide (2-FAA, 0.05%) in male Sprague-Dawley rats, and thalidomide (100 mg/kg body weight) was administered intragastrically to intervene in NF-κB activation. The pathological changes in the liver of sacri fi ced rats were assessed after hematoxylin and eosin staining. NF-κB mRNA was ampli fi ed by RT-nested PCR. The alterations of NF-κB and vascular endothelial growth factor (VEGF) expression were analyzed by enzyme-linked immunosorbent assay,immunohistochemistry, and Western blotting.
RESULTS: Rat hepatocytes showed denatured, precancerous,and cancerous stages in hepatocarcinogenesis, with an increasing tendency of hepatic NF-κB, NF-κB mRNA, and VEGF expression, and their values in the HCC group were higher than those in controls (P<0.001). In the thalidomidetreated group, the morphologic changes generated only punctiform denaturation and necrosis at the early or middle stages, and nodular hyperplasia or a little atypical hyperplasia at the fi nal stages, with the expression of NF-κB(χ2=9.93, P<0.001) and VEGF (χ2=8.024, P<0.001) lower than that in the 2-FAA group.CONCLUSION: NF-κB is overexpressed in hepatocarcinogenesis and antiangiogenic treatment down-regulates the expression of NF-κB and VEGF, and delays the occurrence of HCC.
Author Af fi liations: Department of Diagnostics, Medical College (Dong ZZ), Research Center of Clinical Medicine (Yao DF, Wu W, Qiu LW, Sai WL and Yang JL), Department of Laboratory Science (Yao M), and Department of Oncology (Shen JJ and Yao NH), Af fi liated Hospital of Nantong University,Nantong 226001, China; Department of Chemotherapy, Nantong Cancer Hospital, Nantong 226007, China (Yu HB)
Deng-Fu Yao, MD, PhD, Research Center of Clinical Medicine, Af fi liated Hospital of Nantong University, Nantong 226001, China(Tel: 86-513-85052413; Fax: 86-513-85052523; Email: yaodf@ahnmc.com)
© 2010, Hepatobiliary Pancreat Dis Int. All rights reserved.
December 3, 2009
Accepted after revision March 6, 2010
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Protective effects of MCP-1 inhibitor on a rat model of severe acute pancreatitis
- Extrahepatic right hepatic duct diverticulum:a rare entity
- MRI shows clodronate-liposomes attenuating liver injury in rats with severe acute pancreatitis
- Preoperative assessment of hilar cholangiocarcinoma: combination of cholangiography and CT angiography
- Surgical therapy and prognosis of sarcomatoid carcinoma of the gallbladder
- Small-duct primary sclerosing cholangitis with hepatocellular carcinoma requiring liver transplantation