从头算分子动力学研究O-与CH3F反应的产物通道
2010-11-06吴琍霞戴静华周晓国刘世林
吴琍霞 于 锋 刘 静 戴静华 周晓国 刘世林
(合肥微尺度物质科学国家实验室(筹),中国科学技术大学化学物理系,合肥 230026)
从头算分子动力学研究O-与CH3F反应的产物通道
吴琍霞 于 锋 刘 静 戴静华 周晓国*刘世林
(合肥微尺度物质科学国家实验室(筹),中国科学技术大学化学物理系,合肥 230026)
在B3LYP/6-31+G(d,p)理论水平下采用基于波恩-奥本海默近似的从头算分子动力学方法重新研究了O-与CH3F反应经抽氢生成OH-和生成H2O的两条产物通道.反应轨线从反应初始过渡态开始,采用300 K时的热取样确定初始条件,同时为对比不同的初始碰撞平动能条件下产物通道的变化,分别限定过渡矢量上的能量为2.1、36.8及62.8 kJ·mol-1进行轨线计算,所有轨线计算的结果表明抽氢生成OH-的过程始终为主要的产物通道.我们的计算不仅进一步证实了以往实验的结论,而且描绘了抽氢生成OH-和生成H2O这两个产物通道在反应出口势能面上的动态反应路径,更为深刻地揭示了该反应的微观机理.
从头算分子动力学;氧负离子;氟甲烷;反应机理;过渡态
负离子与分子的反应在大气化学、燃烧化学、有机化学等研究领域中占有重要的地位[1-4],深入了解这些反应的机理有助于我们更清楚地认识离子-分子化学反应的本质,并为探索液相化学反应过程提供重要的线索.氧负离子(O-)是一种高反应活性的负离子,它所参与的化学反应是有机含氧负离子的最直接来源,在有机化学、电离层大气化学、质谱探测分析等诸多领域都有大量涉及,因此吸引了实验工作的广泛研究[5].
在众多的相关反应中,O-和卤代烷烃的反应作为典型代表受到了格外的关注.Futrell和Tiernan[6]利用串联质谱研究了在较低的碰撞平动能条件下O-与CH3F的反应,由测量得到的反应速率常数推知产物离子的分布约为OH-(82%)和CHF-(18%). Tanaka等[7]在流动余辉装置中测量得到的产物离子分支比为OH-占98%、CHF-和F-均占1%.Dawson等[8]在离子回旋共振质谱实验中观测得到的结果则为OH-占80%、CHF-占20%.Peverall等[9]则采用离子选择流动管研究了300 K和66.67 Pa条件下O-和O-2与卤代烷烃分子的反应,获得了反应速率常数和产物通道分支比等信息,其中O-与CH3F的反应速率常数为1.4×10-9cm3·molecule-1·s-1,离子产物分别是OH-(68%)、CHF-(31%)和F-(1%).显然,生成OH-和CHF-离子的过程是该反应的主要产物通道,而生成F-的反应通道分支比非常小.但是,不同实验观测的结果也存在着一些差异,而产生差异的原因被认为是由于实验条件和负离子探测手段的不同而造成的,其中最重要的因素可能是在流动管等实验研究过程中普遍存在的二次反应的影响[5].例如,在O-与CH3F的反应过程中就可能发生初生产物OH-与CH3F的反应,进而影响OH-和F-这两种负离子产物的分支比.
由于质谱实验仅能给出离子产物的部分信息,因此真实的化学反应过程显然无法从这些有限的信息中得到准确的描述.相对而言,高水平的量子化学理论计算则能够为澄清反应机理提供全面的信息,同时也能够证实实验观测结果的合理性.这样,开展该反应的理论研究显然对于揭示其反应机理、深入理解分子-离子作用机制十分必要.事实上, Yamamoto等[10]就曾采用密度泛函理论(DFT)计算了O-与CH3F反应的部分势能面特征,试图阐明实验观测的离子产物生成机理.针对这个反应中可能存在的如下三个产物通道:

Yamamoto等分别在B3LYP/6-31+G(d,p)和B3LYP/ aug-cc-pVTZ理论水平下优化了反应势能面上中间体和过渡态的分子结构,结合内禀反应坐标(IRC)理论计算给出了该反应的势能剖面和反应机理.此外,通过比较O-与CF4、CHF3、CH2F2和CH3F的反应速率常数,他们认为离子偶极相互作用是决定O-与这类分子发生碰撞反应时相互作用的关键因素.然而,他们未能在此基础上进一步研究该体系的动力学反应特征.
最近,我们分别利用IRC和基于波恩-奥本海默近似的分子动力学(Born-Oppenheimer molecular dynamics,BOMD)理论计算对比研究了该反应体系SN2产物通道的静态和动态反应路径,揭示了一条全新的动力学反应产物通道,即生成HF和CH2O-的过程[11].有意思的是,通过对静态和动态反应路径的比较,我们发现加在该反应体系原子核上的平动能大小对动力学反应路径产生决定性的影响,因此像Yamamoto等单纯靠寻找最小能量反应途径的方法来研究反应机理是远远不够的.更深入的计算还表明,SN2过渡态的过渡矢量与其余“实”振动模式之间的耦合强弱是决定生成F-+CH3O还是生成HF+CH2O-产物通道的根本因素.
相对分支比小的SN2产物通道,抽氢生成OH-和生成H2O+CHF-的过程是该反应的主要产物通道,因此开展其相关的反应动力学研究对于全面了解反应机理、判断实验观测结果的合理性具有更为重要的作用.为此,我们采用从头算分子动力学方法重新研究了这两个产物通道,并且通过改变加在反应初始过渡态的过渡矢量上的能量来模拟在不同碰撞条件下的反应情形.最终,通过与Yamamoto等反应势能剖面的比较能够更为深刻地揭示了该反应的微观反应机理,并为进一步研究O-与其他有机分子的反应机理提供了参考.
1 计算方法
本文的计算工作均在Gaussian 03程序包[12]中完成.首先采用密度泛函理论中的B3LYP方法[13-14],在6-31+G(d,p)水平下优化了O-离子进攻CH3F中氢原子的过渡态分子构型,即Yamamoto等[10]获得的反应势能面上对应抽氢生成OH-和生成H2O+ CHF-的过程的初始过渡态TS1.同时,为了研究不同方法和基组水平对该过渡态优化构型可能存在的影响,采用二阶微扰MP2方法[15-16]及较大的基组aug-cc-pvTZ[17]重新进行构型优化,从而对比该过渡态在B3LYP/aug-cc-pvTZ,MP2/6-31+G(d,p)及MP2/ aug-cc-pvTZ水平下的优化构型.最终,从B3LYP/6-31+G(d,p)水平下获得的TS1的优化构型开始,在相同计算水平下进行BOMD计算[18-20].具体计算过程中采用了预测-校正算法,并在计算新的解析Hessian矩阵(能量的二阶偏导数矩阵)前在5个梯度点上更新Hessian[21-22].动力学轨线计算的初始条件由300 K温度下的热取样决定[23-24],即针对TS1的振动取样温度和转动取样温度均设为300 K,该温度与前人实验条件[9]相符.计算过程中,轨线的步长设定为0.25 amu1/2·bohr,而对于每一条轨线我们设定的最大计算步数为5000.当生成的产物很好地分离(即不同产物之间的质心距离超过13 bohr)或者计算的总步数超过5000时,轨线计算将自动终止.
除了正常的热取样确定初始条件外,为了模拟O-与CH3F在较低、中等和较高的初始碰撞平动能条件下产物通道的变化,我们还对加在TS1虚频振动模式即过渡矢量(transition vector)上的能量加以限定比较.具体计算中,分别采用2.1、36.8及62.8 kJ·mol-1平动能代表反应物在较低、中等和较高碰撞能的初始条件.
一般来讲,要获得多通道化学反应的精确通道分支比至少需要计算几百条反应轨线.如果能够拟合得到反应的全维势能面,那么实现大量轨线的计算是可行的.然而,对O-+CH3F的反应体系而言,构建其全维反应势能面将非常困难.这主要是因为以下三个原因:首先,在该体系势能面上的一些区域自恰场计算极难收敛;其次,由于六原子反应体系,自由度较多,构建高精度的全维势能面计算量非常大;更为重要的是,由于对该类反应体系的作用机制并不明确,因此缺乏恰当的拟合函数.在这种情况下,采用BOMD理论方法可以避开全维反应势能面的构建,大大节省了计算资源.我们前期的理论计算工作[11]也证实了其结果的可靠性.当然,即便如此这种动力学计算同样需要非常大的计算量.考虑到计算时间和轨线计算过程中的自恰场计算收敛的问题,我们总共计算了97条轨线.其中,利用正常的热取样确定初始条件,不对分配在过渡矢量上的能量做任何限定条件下,总共计算了57条轨线,用以评估实际反应中的主要产物通道;当限定在过渡矢量上的能量为2.1 kJ·mol-1时,计算了10条轨线,而限定在过渡矢量上的能量为36.8和62.8 kJ·mol-1时,则分别计算了15条轨线,由此对比反应在不同的初始碰撞平动能条件下产物通道的变化.需要说明的是,尽管当前的计算中仅涉及有限数目的反应轨线,但是如下文所示,我们的结果依然和实验结果符合得很好.
此外,轨线计算所得产物的电荷分布由Mulliken电荷布居分析[25]确定.我们以往的理论工作[11,26-28]也证实了该方法可以很好地描述离子-分子反应中产物的电荷分布特征.本文工作中,我们以轨线达到停止标准时的分子结构为研究对象,对该分子结构分别做单点能量计算和Mulliken电荷布居分析,根据获得的负电荷分布状况确定最终的离子产物和中性碎片.
2 结果与讨论
2.1 抽氢反应通道和生成水反应通道的入口势能面特征
在离子诱导偶极作用下,当O-靠近CH3F的氢原子端时可迅速形成离子络合物CH3F…O-,之后经初始过渡态TS1发生抽氢过程,生成离子偶极络合物OH-…CH2F.图1显示了在B3LYP/6-31+G(d,p), B3LYP/aug-cc-pvTZ,MP2/6-31+G(d,p)及MP2/augcc-pvTZ水平下优化得到的TS1分子构型.显然,各理论水平下分子优化构型没有明显差异,这表明当前反应途径中,采用B3LYP/6-31+G(d,p)方法来处理是合理的.在B3LYP/6-31+G(d,p)获得的优化构型中,C和H以及O和H之间的距离分别为0.1365和0.1146 nm,而O、H和C之间的夹角为151.79°.
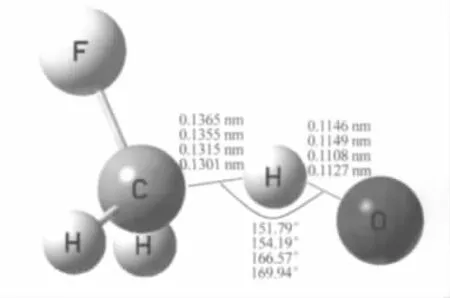
图1 O-+CH3F反应的初始阶段过渡态TS1的优化分子构型Fig.1 Optimized geometry of the initial transition state TS1 of the O-+CH3F reactionThe geometry parameters from the upper to the bottom are obtained at the B3LYP/6-31+G(d,p),B3LYP/aug-cc-pvTZ,MP2/6-31+G(d,p), and MP2/aug-cc-pvTZ levels,.respectively.
正如Yamamoto等[10]的计算所示,抽氢和生成水的反应产物均是由络合物OH-…CH2F经碰撞解离或进一步异构解离得到.具体而言,络合物OH-…CH2F可以经碰撞直接解离,生成OH-+CH2F(即抽氢产物通道);络合物OH-…CH2F还可经另一过渡态异构为H2O与CHF-形成的离子偶极络合物H2O…CHF-,继而碰撞解离生成产物H2O和CHF-.
2.2 抽氢反应和生成水产物通道的反应轨线总结
从反应入口的初始过渡态TS1开始,在正常的热取样确定初始条件,并不对分配在过渡矢量上的能量做任何限定的条件下,我们总共计算了57条轨线,如表1所示.计算获得的轨线中总能量变化在0.16-7.33 kJ·mol-1范围内,始终低于化学精度(8.4 kJ·mol-1),而总的角动量变化则在10-9ħ-10-7ħ(ħ为约化普朗克常量)范围.尽管通过减小计算步长可以改善能量守恒的情况,但是计算量会相应大大增加,因此我们只需控制反应过程中能量守恒满足化学精度要求即可.此外,考虑到该反应是在基态双重态势能面上发生的,我们也考查了轨线计算过程中的自旋污染情况.其中对于该双重态体系而言,投影前自旋角动量平方〈S2〉的本征值接近0.75,这说明自旋污染并不严重,当前多选取的B3LYP/6-31+G(d,p)计算水平是合适的.最终的反应产物由Mulliken电荷布居分析可知,分别对应OH-+CH2F和H2O+ CHF-产物,而不是OH+CH2F和H2O+CHF.
在这57条轨线中,35条轨线对应生成的产物是OH-+CH2F,6条轨线对应生成H2O+CHF-产物,2条轨线显示的产物是CH2OH+F-,14条轨线未在5000步时完成产物分离,停留在离子-分子络合物状态.当然,随着反应步数的进一步增加,这14条轨线中的离子-分子络合物最终分离,分别对应OH-+ CH2F和H2O+CHF-产物.
为了对比反应在不同的初始碰撞平动能条件下产物通道的变化,我们在热取样确定初始条件的前提下,分别限定在过渡矢量上的能量为2.1 kJ·mol-1时,计算了10条反应轨线,而限定在过渡矢量上的能量为36.8和62.8 kJ·mol-1时各自计算了15条轨线.在这总共40条轨线中,大部分轨线的总能量变化低于4.0 kJ·mol-1,其中能量守恒最差的一条轨线能量变化达到8.0 kJ·mol-1,但仍然在化学精度的范围内.总的角动量变化始终在10-8ħ左右.
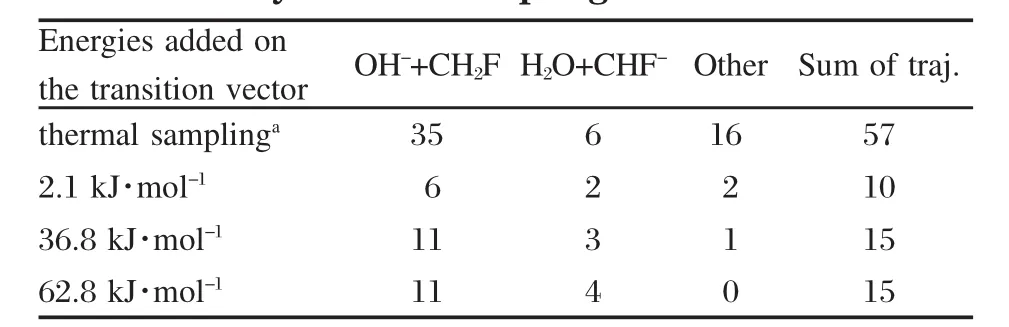
表1 初始条件由300 K热取样确定的条件下O-与CH3F的反应轨线总结Table 1 Trajectory summary for the reaction of O-with CH3F,with the starting conditions determined by thermal sampling at 300 K
当限定在过渡矢量上的能量为2.1 kJ·mol-1时, O-与CH3F的反应近似是在热碰撞条件下发生,相应的相对平动能非常小.在我们计算的10条轨线中,6条轨线最终生成产物OH-+CH2F,即抽氢反应通道;2条轨线最终生成产物H2O+CHF-,即生成H2O的反应通道;一条轨线在5000步时停留在H2O…CHF-中间体络合物,在增加反应时间后最终可以解离为H2O+CHF-;此外,还有一条轨线生成了产物HF+CH2O-,这显然是由于计算过程中反应轨线耦合到别的产物路径上去了.
当加在过渡矢量上的能量为36.8 kJ·mol-1时,我们计算的15条反应轨线中有11条生成产物OH-+CH2F,3条生成H2O+CHF-产物,另有1条轨线由于意外的耦合生成产物F-+CH2OH.而当加在过渡矢量上的能量为62.8 kJ·mol-1时,计算的15条反应轨线中11条对应抽氢反应过程,生成OH-+CH2F产物,其余4条轨线对应生成水的产物通道,得到H2O+CHF-的产物.
当前计算的所有轨线及相应的产物归纳在表1中.显然,在300 K热取样条件下,O-+CH3F反应中通过抽氢过程生成OH-+CH2F产物为主要通道,而且即便是考虑到加在初始反应过渡态TS1的过渡矢量上能量变化,抽氢生成OH-离子的产物通道始终占据主要地位.此外,从我们有限数量的轨线计算来看,加在初始反应过渡态TS1的过渡矢量上能量增加,并没有使抽氢反应或生成水的产物通道分支比有明显的上升.
2.3 抽氢反应通道和生成水反应通道的动态反应路径
通常,动态反应通道与静态反应通道在产物端会有所差异.当前计算所涉及的抽氢产物通道和生成水的产物通道也是如此,其对应的静态反应途径在前面已简要说明,而动态反应过程则略有不同.
图2列举了我们计算得到的一条典型抽氢产物通道的反应轨线结果,及反应体系相对势能随时间的变化.反应经历最初的过渡态TS1后,迅速抽氢形成OH-…CH2F中间体络合物.之后在络合物解离的过程中,在强烈的离子诱导偶极作用下OH-围绕CH2F运动持续一段时间,期间甚至形成弱的氢键,最终生成OH-和CH2F.
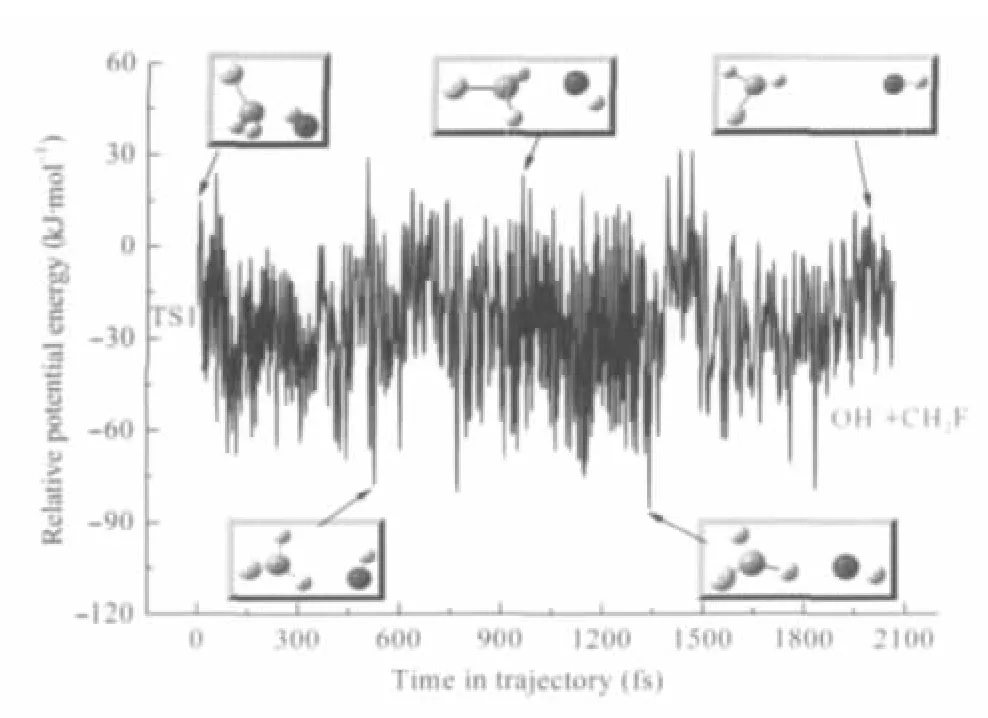
图2 生成OH-+CH2F产物的典型反应轨线中势能随时间变化的曲线Fig.2 Potential energy profile for a typical reaction trajectory leading to OH-+CH2F
类似地,生成H2O+CHF-产物的典型反应轨线中随时间变化的势能曲线见图3.显然,该产物通道的实现是分步进行的.起初的过程与前述的抽氢通道相似,反应在经历最初的过渡态TS1后,迅速抽氢形成OH-…CH2F中间体络合物.之后OH-在围绕CH2F运动时迅速夺取CH2F上的一个质子形成另一种中间体络合物H2O…CHF-,随后在新的络合物解离过程中,同样在离子诱导偶极作用下CHF-和H2O分子相伴运动一段时间,最终生成CHF-和H2O.对比这种动态反应机理,其过程与基于势能剖面的反应机理(静态反应途径)[10]是一致的.
由于当前计算中轨线数目相对较少,很难对涉及的中间体络合物的寿命进行统计分析.然而,在分析具体的反应轨线时,我们发现当过渡矢量上的能量为62.8 kJ·mol-1时,有一条生成OH-+CH2F的轨线仅仅用了大约310 fs的时间就完成了,说明反应过程中OH-…CH2F中间体络合物的寿命极其短暂,甚至可以认为是从过渡态TS1直接解离生成产物OH-+CH2F的.
2.4 与实验结果的比较
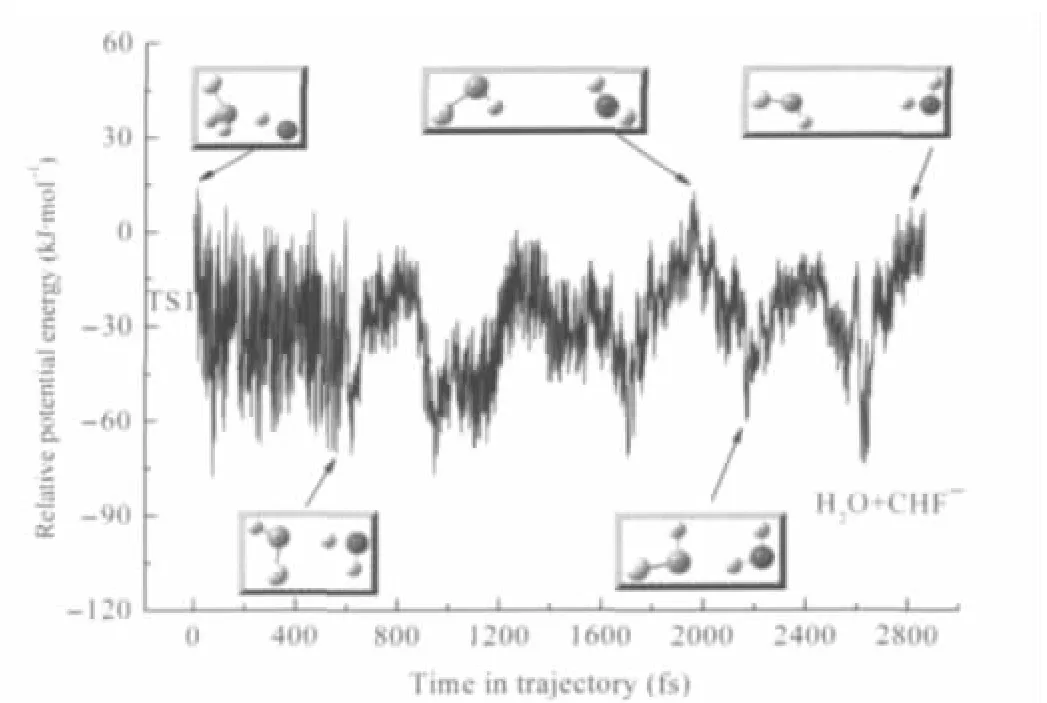
图3 生成H2O+CHF-的典型反应轨线中势能随时间变化的曲线Fig.3 Potential energy profile for a typical reaction trajectory leading to H2O+CHF-
仔细对比以往的实验研究,Futrell和Tiernan[6]在串联质谱中观测的产物分布为OH-约占82%、CHF-约占18%,而Tanaka等[7]在流动余辉实验中给出的产物分支比为OH-占98%、CHF-和F-均占1%.考虑到两者的实验条件非常类似,均是由近300 K温度下热碰撞引起的化学变化,因此两实验中O-与 CH3F反应的初始碰撞平动能均较低.然而由于负离子探测效率的不同,两实验测量得到的CHF-产物分支比存在较大差异.此外,Dawson等[8]测量得到的产物分布为OH-占80%、CHF-占20%;Peverall等[9]观测的反应负离子产物主要有OH-(占68%)、CHF-(占31%)和F-(占1%).尽管这些实验结果彼此之间存在较大的差异,而且实验测量具有较大的误差,如Peverall等[9]报道的实验误差约为20%,但是所有的实验研究[6-9]都证实抽氢生成OH-+CH2F是最主要的产物通道,这显然与我们当前的计算结果吻合得很好.这样,我们的计算将为进一步研究O-与有机分子的反应机理提供了有价值的参考.
3 结论
我们在B3LYP/6-31+G(d,p)理论水平下采用BOMD方法研究了O-与CH3F反应经抽氢生成OH-和生成H2O的两个产物通道.通过对比静态反应途径和动态反应通道的差别,我们详细描绘了抽氢生成OH-和生成H2O这两个产物通道在反应出口势能面上的动态反应路径,更为深刻地揭示了该反应的微观机理,并且证实抽氢生成OH-离子是该反应的主要产物通道,这与以往实验测量的结论完全一致.
此外,我们还通过改变加在反应初始过渡态的过渡矢量上的能量来模拟在不同碰撞条件下的反应轨线.反应轨线依然从初始过渡态TS1开始,当加在过渡矢量上的能量分别为2.1、36.8以及62.8 kJ· mol-1时,抽氢生成OH-离子的过程始终表现为主要的产物通道,这进一步证实了以往实验的结论.对该反应体系的计算将为深入研究O-与有机分子的反应机理提供依据和参考.
1 Bowers,M.T.Gas phase ion chemistry.Vol.1.New York: Academic Press,1984:45-82
2 Wayne,R.P.Chemistryofatmosphere.Oxford:Clarendon Press, 1991:225-256
3 Fialkov,A.B.Prog.Energy Combust.Sci.,1997,23:399
4 DePuy,C.H.J.Org.Chem.,2002,67:2393
5 Lee,J.;Grabowski,J.J.Chem.Rev.,1992,92:1611,and references therein
6 Futrell,J.H.;Tiernan,T.O.Ion molecule reactions.Vol.2. Franklin,J.L.Ed.London:Butterworths,1972:485-551
7 Tanaka,K.;Mackay,G.I.;Payzant,J.D.;Bohme,D.K.Can.J. Chem.,1976,54:1643
8 Dawson,J.H.J.;Jennings,K.R.J.Chem.Soc.Faraday Trans 2, 1976,72:700
9 Peverall,R.;Kennedy,R.A.;Mayhew,C.A.;Watts,P.Int.J.Mass Spectrom.Ion Processes.,1997,171:51
10 Yamamoto,M.;Yamashita,K.;Sadakata,M.J.Mol.Struct.-Theochem,2003,634:31
11 Yu,F.;Wu,L.X.;Liu,S.L.;Zhou,X.G.J.Mol.Struct.-Theochem,2010,947:1
12 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Reversion B.05.Pittsburgh,PA:Gaussian Inc.,2003
13 Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B,1988,37:785
14 Becke,A.D.J.Chem.Phys.,1993,98:5648
15 Møller,C.;Plesset,M.S.Phys.Rev.,1934,46:618
16 Frisch,M.J.;Head-Gordon,M.;Pople,J.A.Chem.Phys.Lett., 1990,166:281
17 Kendall,R.A.;Dunning,T.H.;Harrison,R.J.J.Chem.Phys., 1992,96:6796
18 Thompson,D.L.Modern methods for multidimensional dynamics computations in chemistry.Singapore:World Scientific,1998: 143-189
19 Schlegel,H.B.J.Comput.Chem.,2003,24:1514
20 Schlegel,H.B.Bull.Korean Chem.Soc.,2003,24:837
21 Millam,J.M.;Bakken,V.;Chen,W.;Hase,W.L.;Schlegel,H.B. J.Chem.Phys.,1999,111:3800
22 Bakken,V.;Millam,J.M.;Schlegel,H.B.J.Chem.Phys.,1999, 111:8773
23 Schleyer,P.v.R.;Allinger,N.L.;Clark,T.;Gasteiger,J.; Kollman,P.A.;Scheafer III,H.F.;Schreiner,P.R.Encyclopedia of computational chemistry.Chichester:Wiley,1998:402-407
24 Peslherbe,G.H.;Wang,H.;Hase,W.L.Adv.Chem.Phys.,1999, 105:171
25 Mulliken,R.S.J.Chem.Phys.,1955,23:1833
26 Yu,F.;Zhao,Y.G.;Wang,Y.;Zhou,X.G.;Liu,S.L.Acta Chim. Sin.,2007,65:899 [于 锋,赵英国,王 勇,周晓国,刘世林.化学学报,2007,65:899]
27 Wang,X.L.;Yu,F.;Xie,D.;Liu,S.L.;Zhou,X.G.Acta Chim. Sin.,2008,66:2499 [王新磊,于 锋,谢 丹,刘世林,周晓国.化学学报,2008,66:2499]
28 Wang,J.X.;Yu,F.;Liu,J.;Liu,S.L.;Zhou,X.G.Acta Phys.-Chim.Sin.,2008,24:1393 [王俊霞,于 锋,刘 静,刘世林,周晓国.物理化学学报,2008,24:1393]
Ab initio Molecular Dynamics Investigation on the Production Channels for the Reaction of O-with CH3F
WU Li-Xia YU Feng LIU Jing DAI Jing-Hua ZHOU Xiao-Guo*LIU Shi-Lin
(Hefei National Laboratory for Physical Sciences at the Microscale,Department of Chemical Physics, University of Science and Technology of China,Hefei230026,P.R.China)
H-atom abstraction and H2O production channels for the reaction of O-with CH3F were reinvestigated using the ab initio molecular dynamics method at the B3LYP/6-31+G(d,p)level of theory and based on the Born-Oppenheimer approximation.The reactive trajectories were initiated at the transition state of H-atom abstraction. Thermal sampling at 300 K was chosen to determine the initial conditions.Additionally,the energies added to the transition vector of the barrier were restricted to 2.1,36.8,and 62.8 kJ·mol-1,separately,to reveal the impact of different initial collision energies on the reaction pathways.The results of all the trajectory calculations demonstrate that the H-atom abstraction channel is the dominant production channel.Therefore,our calculations are consistent with previous experimental conclusions.Furthermore,the dynamic reaction pathways for H-atom abstraction and the H2O production channels on the exit-channel potential energy surface are described based on our calculations and thus a comprehensive reaction mechanism is revealed at the microscopic level.
Ab initio molecular dynamics;Atomic oxygen radical anion;Methyl fluoride;Reaction mechanism; Transition state
O643
Received:April 12,2010;Revised:May 24,2010;Published on Web:July 9,2010.
*Corresponding author.Email:xzhou@ustc.edu.cn;Tel/Fax:+86-551-3600031.
The project was supported by the National Natural Science Foundation of China(20603033,10979042)and National Key Basic Research Program of China(973)(2007CB815204).
国家自然科学基金(20603033,10979042)和国家重点基础研究发展规划项目(973)(2007CB815204)资助
ⒸEditorial office of Acta Physico-Chimica Sinica
