微乳液成核-离子交联制备阿司匹林/壳聚糖纳米微球及其体外释放行为
2010-11-06金淑萍何文龑韩玉琦魏玉娟
金淑萍 冯 雷 何文龑 韩玉琦 魏玉娟
(河西学院化学系,西部资源环境化学重点实验室,甘肃张掖 734000)
微乳液成核-离子交联制备阿司匹林/壳聚糖纳米微球及其体外释放行为
金淑萍 冯 雷*何文龑 韩玉琦 魏玉娟
(河西学院化学系,西部资源环境化学重点实验室,甘肃张掖 734000)
以自制阿司匹林为药物模型,壳聚糖(CS)为载体源,采用微乳液成核-离子交联法制备了阿司匹林/壳聚糖纳米缓释微球.分别用傅里叶变换红外(FTIR)光谱、场发射扫描电子显微镜(FESEM)、透射电子显微镜(TEM)、动态激光光散射(DLLS)、X射线粉末衍射(XRD)等表征了纳米微粒的化学组成、外观形貌、平均粒径和粒径分布、微球中壳聚糖的晶体结构以及阿司匹林的分布形态.结果表明,利用微乳液成核-离子交联法制备的阿司匹林/壳聚糖微球平均粒径约为88 nm且粒径分布均匀,成核后壳聚糖结晶形态基本未变,阿司匹林以分子形态分布于微粒中,分子间未形成堆砌,为无定形态.采用UV-Vis分光光度计考察了微球的药物包封率、载药量,并对微球在生理盐水和葡萄糖溶液中的释药行为进行跟踪.结果表明,微球的载药量可达55%,药物包封率可达42%,实验条件下具有较好的药物缓释作用.
壳聚糖; 纳米微球; 微乳液成核; 药物缓释; 构象
近年来,在医学和制药学领域,人们一直在寻求一种低环境负荷、低能耗、低成本的制造高度功能化纳米胶囊的方法.期望纳米胶囊可以将各种物质包裹在其内核,且努力使它们的尺寸与典型病毒尺寸相近,使其能够在血液中长时间循环[1-2],并最终穿透肿瘤附近组织被破坏的毛细血管,达到肿瘤靶向治疗的目的[3-4],实现对肿瘤组织的“被动靶向性”.因而纳米微粒的制备和性能研究是当前药物递送系统的研究热点[5-7].从体内安全性的角度考虑,人们逐渐寄希望于以天然高分子材料为原料开发聚合物纳米胶囊.壳聚糖(CS)由于具有抗溃疡、防止细菌侵蚀、可生物降解及良好的生物相容性等特性[8-10],近年来得到了越来越多的关注.壳聚糖基纳米微粒可使药物分子顺利通过上皮组织,促进药物的渗透吸收[8,11].Tanima等[12]用交联法制备了超细壳聚糖纳米粒子.经静脉注射,纳米粒子可避开网状类皮的吞噬,在血液中长时间保留,用放射性元素(99m Tc)示踪壳聚糖纳米粒子在老鼠体内的分布,2 h后仍可在血液中检测到,同时粒子还分布到心脏、肝脏、肾脏、膀胱、脊柱和骨骼中,说明这种纳米粒子可用于骨骼的成像和靶向传输.以上研究证实合成稳定、可再生并能负载和包封药物的壳聚糖纳米微粒的可行性及必要性.
具有负载、靶向、控释等作用的壳聚糖纳米微球的制备方法有乳化交联、溶剂蒸发、形成复乳、喷雾干燥、沉淀析出及复凝聚法等.这些方法需在较苛刻的制备条件如存在有机溶剂、乳化剂和超声振荡下进行,因而在实际应用上受到了一定程度的限制[13].
阿司匹林是一种弱酸性药物,遇湿气即缓慢水解成为水杨酸,主要用作解热镇痛药和抗风湿药.研究表明:小剂量的阿司匹林具有较好的抗血栓作用,可用于心血管系统疾病的预防和治疗.但普通的阿司匹林片,在体内水解成水杨酸后对胃肠道黏膜有刺激作用.为了减少患者的服药次数,降低胃肠道的不良反应,使血液中药物浓度平稳持久,提高药物疗效,可将其制成缓释微球并通过静脉注射使用.
本文利用壳聚糖分子在不同pH值的水溶液中构象的变化,采用一种介于沉淀析出法和溶液中离子交联法的新方式,可称之为微乳液成核-离子交联法,使壳聚糖从乳液中以离子交联纳米微粒的形式析出,同时,通过氢键相互作用和疏水相互作用,将溶液中的阿司匹林和水杨酸(阿司匹林的水解产物)包埋到微粒之中,制备得到阿司匹林/壳聚糖纳米微球,用FTIR、FESEM、TEM、DLLS和XRD对其结构和形貌进行了表征,考察了微粒在注射用生理盐水和葡萄糖溶液中的释药行为,为开发利用可静脉注射型壳聚糖基纳米微粒奠定理论基础.
1 实验方法
1.1 原料及试剂
壳聚糖,中国浙江金壳生物化学有限公司,脱乙酰度为90.8%,动力黏度为142 mPa·s,平均聚合度约为350;阿司匹林(实验室自制),用作药物模型; Span-80,广州市润华食品添加剂有限责任公司精细化工厂,食品级;Tween-80,佛山市科的气体化工有限公司,食品级;柠檬酸三钠及其它试剂均为分析纯,未经纯化直接使用.
1.2 阿司匹林/壳聚糖纳米微球的制备
称取0.20 g壳聚糖于250 mL单口圆底烧瓶中,加入2%(质量分数)醋酸溶液250 mL,开启电动搅拌装置缓慢搅拌直至壳聚糖完全溶解.边搅拌边加入Span-80 0.4 mL和Tween-80 0.6 mL,继续搅拌30 min使之形成均匀、透明、稳定的微乳液.接着加入0.26 g经仔细研磨的阿司匹林粉末,高速搅拌使之扩散均匀,然后,缓慢滴加1 mol·L-1的NaOH溶液,同时匀速搅拌,用PHS-3B型精密pH计(中国上海雷磁)在25℃下检测体系pH值的变化,直至微碱性(pH值为7.20).当体系pH值约为4.30时,阿司匹林完全溶解(水解).NaOH溶液滴加完毕后,中速搅拌并加入0.30 g柠檬酸三钠,继续搅拌3 h,使乳液中形成的壳聚糖微球交联固化.得到的透明乳液用离心机离心分离,上层清液用Lambda 35型紫外-可见分光光度计(美国Perkin Elmer)测定299 nm处阿司匹林的吸光度,下层粘度较大的液体经液氮快速冷冻后用冷冻干燥仪(英国Labconco)冷冻干燥得淡黄色粉末.
1.3 药物包埋率
1.3.1 阿司匹林标准浓度曲线的制作
准确称取一定量阿司匹林至100 mL容量瓶中,加去离子水20 mL,溶解后,定容至刻度线,摇匀,配成浓度分别为0.0104、0.0124、0.0144、0.0164、0.0184 mol·L-1的标准溶液.分别取标准溶液用UV-Vis分光光度计测定溶液在299 nm处的吸光度值(A),再以A值对溶液浓度(c)作图,得线性回归方程A=70.045c-0.0432,相关系数r2=0.99696.
1.3.2 阿司匹林/壳聚糖纳米微粒药物包封率的测定
将完全干燥的阿司匹林/壳聚糖淡黄色粉末仔细研磨成极细的粉末,精确称取0.50 g粉末加入50.00 mL的0.1 mol·L-1盐酸溶液中,搅拌24 h,过滤,精确移取5 mL滤液于50 mL容量瓶中,用0.1 mol·L-1盐酸稀释定容至刻度,UV-Vis分光光度计记录溶液在299 nm处的吸光度,通过标准工作曲线线性回归方程计算出阿司匹林的含量,按下列公式计算载药量(L,%)和包封率(R,%)[14].

式中,m1为微球中的阿司匹林质量;m2为称取的微球质量;m3为投入阿司匹林的总量.
1.4 阿司匹林/壳聚糖纳米微粒结构的表征
将冷冻干燥后的淡黄色粉末样品进行如下测试:红外光谱在NEXUS 670 FTIR仪(美国Nicolet)上测定,KBr压片;XRD晶相分析在RU-200B型X射线衍射仪上进行(日本Rigaku公司),Cu Kα辐射,管电流100 mA,管电压40 kV;淡黄色粉末样品表面喷金后,用JSM-6701F SEM型场发射扫描电子显微镜(FESEM)(日本JEOL)对样品形貌进行观察;将样品超声分散配制4×10-5g·mL-1的去离子水溶液, 0.1 μm滤膜过滤,用H-600型TEM和BI-200SM型激光光散射仪观察样品的平均粒径及其分布.
1.5 体外释放实验
模型药物的体外释放实验按如下方法进行:称取50 mg阿司匹林/壳聚糖纳米微粒于50 mL容量瓶中,加入注射用生理盐水或其他释放介质定容后,转移到100 mL的锥形瓶中,封严,在(37.0±0.5)℃的温度范围内,分别于1、2、2、3、3、3、5、5、5、5、10、10、10、10、10、20、20、20、20、20、30、30、30、30、30 min量取4 mL缓释溶液,同时补充4 mL注射用生理盐水或其他释放介质,用UV-Vis分光光度计记录溶液在299 nm处的吸光度值,按线性回归方程计算释放t时刻阿司匹林的累积释放量,并通过下式计算累积释放分数(Freleased):

式中,mt为释放t时刻阿司匹林的累积释放量,m为50 mg样品中阿司匹林的含量.
2 结果与讨论
2.1 溶液pH值变化时壳聚糖分子构象的变化及纳米微粒的形成
聚合物的构象是指由于单键内旋转而导致的聚合物分子链上的原子和基团在空间的不同排列.壳聚糖是一种多功能的天然阳离子聚电解质,在溶液中的构象一般分为球形、无规线团和刚性棒状,其构象的变化主要受两类因素的影响:结构参数,如分子量和脱乙酰度;溶液性质,如溶液离子强度、溶剂、溶液的温度和pH值等[15].在壳聚糖分子的分子量、脱乙酰度和溶液温度不变的情况下,改变溶液pH值(同时离子强度也在变化),壳聚糖在溶液中的构象将会发生变化.在酸性溶液中,壳聚糖上的伯氨基(NH2)质子化为NH+3(壳聚糖分子上的大量羟基也会质子化),分子内和分子间存在着较强的静电排斥作用,此时,壳聚糖分子呈现无规线团状(如图1所示).当增加溶液的pH值时(离子强度相应增大),NH+3逐渐解离成NH2,静电排斥作用减小,此时分子内及分子间氢键作用逐渐加强,同时反离子将屏蔽部分未解离的NH+3形成偶极子,分子内和分子间偶极-偶极作用加强,使分子构象收缩蜷曲(图1中的球形).另外,壳聚糖分子内和分子间的氢键作用加强必然导致壳聚糖分子与水分子之间的氢键相互作用减弱,而疏水相互作用对壳聚糖分子的构象转变具有重要的影响.
很多研究者采用荧光光谱[16]、静态光散射[17-18]、动态光散射[19]等对溶液中壳聚糖的聚集行为进行了研究,结果表明壳聚糖在稀溶液中的临界聚集浓度(cac)大约是1.0×10-3g·mL-1.本文中壳聚糖的溶液浓度为0.8×10-3g·mL-1,接近于壳聚糖的临界聚集浓度,因而随着溶液pH值和离子强度的增大,相应增大的除壳聚糖分子内的氢键和偶极-偶极相互作用外,还有分子间的氢键及偶极-偶极相互作用,壳聚糖的分子链构象由无规线团转变为球形,同时发生分子间的团聚现象.
随溶液pH值和离子强度的逐渐增加,壳聚糖分子链由较为伸展的无规线团转变为球形构象,表明分子亲水性降低,疏水相互作用加强,而溶液中存在的Tween-80和Span-80对壳聚糖的球形构象有一定的稳定作用.当溶液中加入柠檬酸三钠时, COO-阴离子与壳聚糖分子链上残留的NH+3之间的静电相互作用进一步交联固化了壳聚糖微球(图2).

图1 单分子链壳聚糖在不同pH值稀水溶液中的分子构象Fig.1 Molecular conformation of chitosan in dilute solution with different values of pH
2.2 阿司匹林/壳聚糖纳米微球的FTIR分析和药物包封率测定
样品中各组分间的相互作用能影响分子中化学键的键长、键的方向及强度,从而导致功能基频率及强度的改变.因此,各官能团对红外辐射的吸收位置可能会发生较大程度的偏移.
壳聚糖的FTIR光谱如图3中谱线a所示. 1653.06 cm-1处较强的吸收峰为酰胺I带(C═O的伸缩振动吸收),1599.85 cm-1处的吸收峰为脱乙酰后NH2的面内弯曲振动吸收,而1310.39及1269.48 cm-1处的弱吸收可以归属为酰胺III带(分别为缔合态、游离态未脱乙酰C—N键伸缩振动吸收峰).多糖结构的特征吸收带出现在1156.24 cm-1(吡喃环中C—O—C的不对称伸缩振动峰),1424.92 cm-1(CH2—OH中O—H的面内弯曲振动特征吸收峰), 1082.51 cm-1(CH2—OH中C—O伸缩振动吸收峰)以及1029.98 cm-1(CH—OH中C—O伸缩振动吸收峰)[20-22].1385.28 cm-1处的吸收可能是由于CH的弯曲振动所致.约3423 cm-1处宽而强的吸收峰可归属于糖环上氢键缔合O—H、N—H的伸缩振动.
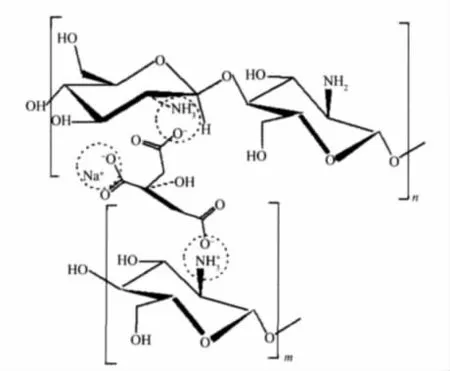
图2 柠檬酸三钠交联壳聚糖的结构示意图Fig.2 Schematic diagram of the structure of chitosan crosslinked by trisodium citrate
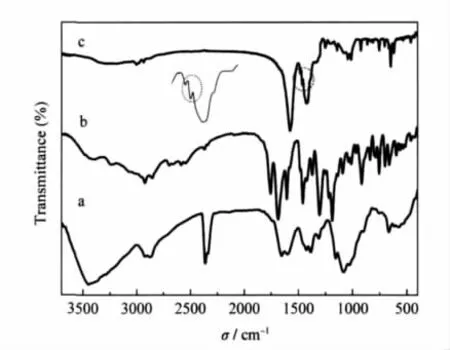
图3 壳聚糖(a)、阿司匹林(b)和阿司匹林/壳聚糖(c)纳米微粒的FTIR光谱图Fig.3 FTIR spectra of the chitosan(a),aspirin(b), and aspirin/chitosan(c)nanosphere
阿司匹林/壳聚糖纳米微球的FTIR光谱如图3中谱线c所示,由于制备过程为微碱性,壳聚糖水解脱乙酰,1653.06 cm-1处酰胺 I带、1310.39及1269.48 cm-1处的酰胺III带相对酰胺II带减弱[23].而1599.85 cm-1处酰胺II带向低波数1575.76 cm-1漂移且强度加强,这可能是由于解离NH2与水杨酸中OH之间的氢键相互作用以及部分未解离NH+3与柠檬酸三钠中COO-之间的静电相互作用使吸收峰位置红移,同时与水杨酸苯环的C═C伸缩振动叠加使吸收峰强度增大.而3423 cm-1处O—H和N—H宽而强的吸收峰相对减弱并向低波数漂移,再次表明NH2的质子化和氢键及静电相互作用的存在.由于水解及酸碱中和,谱线b中1755.31 cm-1处阿司匹林酯基中的C═O和1685.96 cm-1处芳香羧酸中C═O的伸缩振动特征吸收在阿司匹林/壳聚糖谱图中消失.而可以用于鉴定苯环是否存在的C═C伸缩振动的四个谱带是1625-1580 cm-1、1590-1570 cm-1、1525-1470 cm-1和1460-1440 cm-1,其中在阿司匹林/壳聚糖谱图中可以明显观察到位于1481.92、1463.67 cm-1处的两个吸收峰,与壳聚糖在1423.05 cm-1的CH2—OH中OH的面内弯曲振动特征吸收峰及1386.78 cm-1处的CH的弯曲振动吸收峰部分重叠,表明壳聚糖纳米微球中水杨酸成分的存在.另一个应用较多可以鉴定苯环取代类型的吸收峰为754.96 cm-1处的峰,为苯环上四个相邻C—H键的伸缩振动吸收峰.水杨酸中COO-基团的不对称与对称伸缩振动吸收峰分别出现在1610-1550 cm-1及1440-1360 cm-1范围内,这也是谱线c中1575.76与1423.27 cm-1处吸收峰较壳聚糖的FTIR谱线a强度增大的另一个原因,进一步说明在微乳液成核-离子交联过程中,壳聚糖从乳液中以离子交联纳米微粒形式析出的同时,通过氢键相互作用及疏水相互作用,溶液中的水杨酸(阿司匹林的水解产物)也被成功包埋到微粒之中,制备得到了阿司匹林/壳聚糖纳米微球.
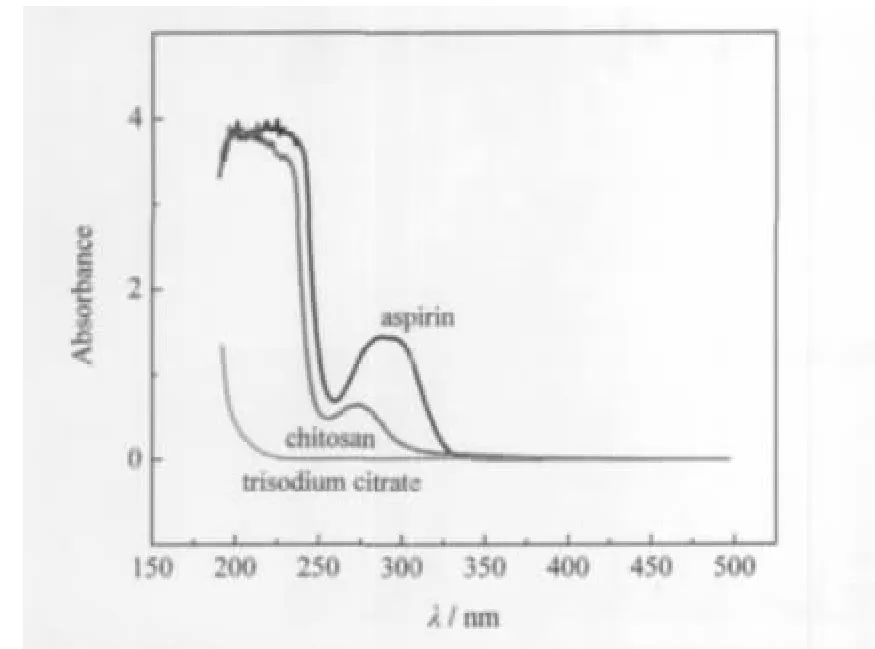
图4 水溶液中阿司匹林、壳聚糖和柠檬酸三钠的UV-Vis光谱图Fig.4 UV-Vis spectra of aspirin,chitosan,and trisodium citrate in aqueous solution
制备过程投料比中阿司匹林的用量约为壳聚糖总量的130%,高速离心后上层清液用UV-Vis分光光度计检测其中所含阿司匹林在299 nm处的吸光度值(图4),代入标准浓度曲线的线性回归方程,求得高速离心后上层清液中阿司匹林的含量约为0.15 g,约占阿司匹林投入量的58%.下层粘度较大的液体经冷冻干燥后得到阿司匹林/壳聚糖淡黄色粉末,研磨后称取0.50 g粉末加入50 mL 0.1 mol·L-1的HCl溶液,搅拌24 h,过滤,稀释,UV-Vis分光光度计记录溶液在299 nm处的吸光度值,通过标准浓度曲线线性回归方程,按公式(1)、(2)计算得载药量约为55%,包封率约为42%.
由于阿司匹林在微碱性溶液中易溶解,导致药物包封率较低.针对微碱性环境中溶解度较小的药物(如抗肿瘤药物紫杉醇)或在微碱性溶液中稳定的氨基酸、肽和蛋白质等新型药物,药物的包封率应会相应提高,而这种微乳液成核-离子交联法制备的壳聚糖纳米微粒为氨基酸、肽和蛋白质的输送提供了新的途径,在方便病人用药和被吸收进入细胞达到细胞内靶的方面具有潜在的价值.
2.3 阿司匹林/壳聚糖纳米微粒的结晶形态及形貌分析
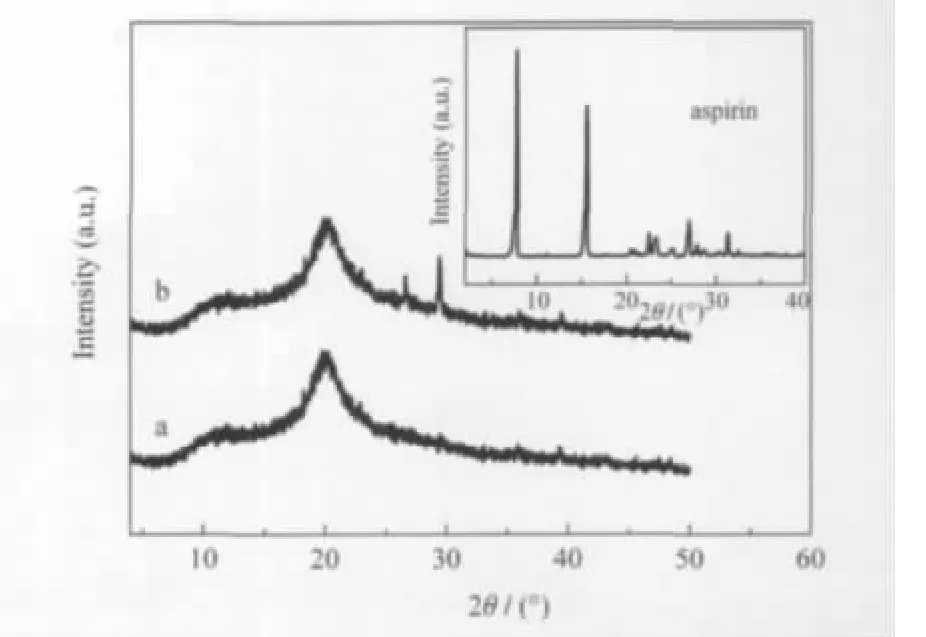
图5 壳聚糖(a),阿司匹林/壳聚糖(b)和阿司匹林(插图)的XRD谱图Fig.5 XRD patterns of chitosan(a),aspirin/chitosan (b),and aspirin(inset)
图5 为壳聚糖和阿司匹林/壳聚糖的XRD谱图.由图5谱线a可以看出,壳聚糖具有一定的结晶性,在2θ为11.9°(100)和20.2°(020)附近有两个较强的基面衍射峰,但衍射峰强度较弱,宽度较大,说明其结晶度较低,与文献报道[24-25]类似.阿司匹林/壳聚糖(图5谱线b)与壳聚糖的XRD谱图基本一致,说明微乳液成核-离子交联过程对壳聚糖的晶体结构和结晶度没有影响,同时也说明阿司匹林对壳聚糖的结晶几乎没有影响.然而,在2θ为26.46°,29.22°处观察到两个弱衍射峰,目前无法将其进行正确解释,推测可能是因为在微乳液成核-离子交联过程中,随着溶液pH值和离子强度的增大,壳聚糖分子内和分子间的氢键和偶极-偶极相互作用也随着增大,而大分子这种长程有序的协同相互作用促使壳聚糖分子链局部排列规整,导致其晶体结构发生微小的变化.图5插图中的谱线显示阿司匹林在衍射角2θ为7.74°和15.52°等处有强的衍射峰[26-27].而这些特征衍射峰在谱线b中没有出现,表明由于氢键和疏水相互作用阿司匹林在壳聚糖载体上高度分散,分子间未形成堆砌,阿司匹林主要以无定形态存在于纳米微粒中.
采用场发射扫描电子显微镜观察未交联壳聚糖和阿司匹林/壳聚糖的形貌特征,结果分别示于图6a和6b.从图6a中可以看出,在碱性环境中,分子内及分子间氢键和偶极-偶极作用加强,促使壳聚糖分子构象收缩蜷曲成球形.但由于未加离子交联剂,粒子表面粗糙.而分子间的氢键及偶极-偶极相互作用使团聚现象发生.而图6b显示的结果表明,利用微乳液成核-离子交联法制得的阿司匹林/壳聚糖呈比较规整的球型,微球的平均直径小于100 nm,较未交联壳聚糖粒子的直径大,而且表面光滑,粒径分布均匀,粘连团聚现象较弱.阿司匹林的加入是粒径增大的主要原因,而阿司匹林和柠檬酸三钠对壳聚糖分子的交联作用可能使粒子表面更为光滑且团聚减轻.

图6 未交联壳聚糖(a)和阿司匹林/壳聚糖(b)的场发射扫描电镜照片Fig.6 FESEM images of uncrosslinked chitosan(a)and aspirin/chitosan(b)
制样过程中的离心分离使溶液浓度局部增大也可能导致团聚,所以在低浓度下可能得到分散状态较好的阿司匹林/壳聚糖微球.将阿司匹林/壳聚糖超声分散到去离子水中,样品在水中的质量浓度为4×10-5g·mL-1,分别用透射电子显微镜和动态激光光散射观察阿司匹林/壳聚糖在去离子水中的分散情况,并测定粒径及粒径分布,结果分别示于图7和图8.从图中可以看出,阿司匹林/壳聚糖颗粒均匀且分散性良好,平均粒径约88 nm.
2.4 阿司匹林/壳聚糖纳米微粒的体外释药性能
壳聚糖分子的构象以及柠檬酸三钠、壳聚糖和阿司匹林的电荷密度与溶液pH值密切相关,因而溶液pH值变化会影响到阿司匹林/壳聚糖纳米微粒的交联密度的变化,从而影响药物释放速率.本文考察了阿司匹林/壳聚糖纳米微粒在pH值分别为1.8和7.2的缓冲溶液中的释放行为,结果表明,在pH值为7.2的缓冲溶液中,阿司匹林/壳聚糖保持收缩状态,药物释放很慢,250 min内释药仅23%.而在pH值为1.8的缓冲溶液中,由于柠檬酸三钠的交联作用消失,壳聚糖分子构象伸展,250 min内药物释放已近90%.与Shu等[28]得到的结果一致.
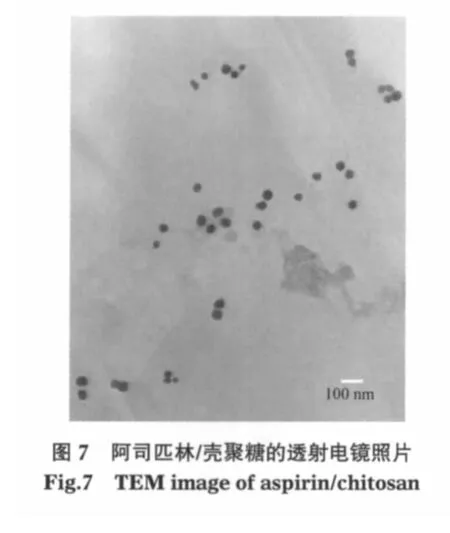
图9所示的是去离子水、注射用生理盐水及葡萄糖溶液中阿司匹林/壳聚糖纳米微粒累积释放量随时间的变化曲线.不难看出,在释放的初始阶段,葡萄糖溶液中阿司匹林/壳聚糖纳米微粒的药物释放速率明显小于生理盐水中释放的速率,40 min内药物释放不到50%,而在生理盐水中释放已达到80%.吸附在微粒表面的药物扩散可能是导致释放初期快速释药的重要因素.经过一个突释阶段后,释放速度减慢,释药约400 min,生理盐水中阿司匹林/壳聚糖纳米微粒释放率约为95%,葡萄糖溶液中释放率约为55%,仍有45%的药物未释放,纳米微球表现出良好的缓释性能.与生理盐水中阿司匹林的释放速率相比,去离子水中阿司匹林/壳聚糖纳米微粒的释放比较缓慢,与葡萄糖溶液中的释放接近.

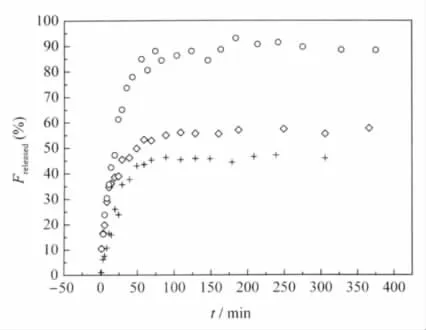
图9 生理盐水(○)、葡萄糖溶液(◇)和去离子水(+)中阿司匹林/壳聚糖纳米微粒的累积释放量随时间的变化曲线(37℃)Fig.9 Release profiles of aspirin/CS nanospheres in saline(○),glucose solution using in medicine(◇)and deionized water(+)at 37℃
研究大分子聚电解质的溶液行为,离子强度是一个关键的影响因素[29].人们已深入地研究了离子强度对壳聚糖溶液性质和构象的影响.Tsaih等[30]对稀溶液中离子强度对壳聚糖分子构象影响的研究发现,随着溶液离子强度的增加,壳聚糖的分子构象从伸展变得蜷曲.在低离子强度时,第三电粘度(the third electroviscous)的影响起主要作用,壳聚糖呈现伸展的分子构象.在高离子强度时,反离子对质子化氨基的屏蔽作用使分子蜷曲.当离子强度增加到无穷大时,所有的质子化氨基都被反离子中和,分子内的静电斥力消失,所有的壳聚糖分子无论分子量大小都变成紧缩的球形,当用脲将分子内的氢键消除时,壳聚糖分子又变得伸展.然而,对于壳聚糖基于静电相互作用和氢键相互作用形成的纳米微球,当溶液中加入小分子电解质(例如NaCl)时,电解质对壳聚糖构象的影响研究较少.本文中图9所示的实验结果表明,NaCl溶液中阿司匹林的释放速率更快,表明在生理盐水中壳聚糖的离子交联密度可能较低,壳聚糖的构象可能由蜷曲的球变化为伸展的蠕虫状链,从而使药物释放.这可能是因为小分子电解质破坏了壳聚糖纳米粒的水合层,由于渗透压的关系由水溶液向粒子内部渗透,与壳聚糖的质子化氨基相互竞争,Na+与柠檬酸盐COO-之间的静电相互作用部分取代了壳聚糖链上NH+3与柠檬酸盐COO-之间的静电相互作用,导致低的交联密度[31].同时,更为重要的是,NaCl的渗入,削弱了壳聚糖分子间和分子内的氢键作用,有可能导致壳聚糖的构象由蜷曲变化为伸展的蠕虫状链,因而使阿司匹林的释放速率由于小分子电解质的加入而加快.可见,小分子电解质对壳聚糖溶液行为的影响,到目前为止,仍有高分子相转变现象、相转变机理、相平衡原理等诸多问题,等待广大高分子工作者进行深入细致的研究.
3 结 论
场发射扫描电子显微镜、透射电子显微镜和动态激光光散射研究结果表明微乳液成核-离子交联法不失为一种制备壳聚糖基纳米载药微球的简单易行而又有效的方法.UV-Vis分光光度计对纳米微球在注射用生理盐水和葡萄糖溶液中的释药行为跟踪结果表明,实验条件下葡萄糖溶液中壳聚糖基纳米载药微球具有良好的药物缓释作用,而生理盐水中较快的释放速率可能归因于小分子盐对壳聚糖分子链中氢键和静电相互作用的破坏.小分子电解质对壳聚糖在溶液中的构象转变的这种影响有待于进一步研究.
1 Yang,Z.;Zheng,S.Y.;Harrison,W.J.;Harder,J.;Wen,X.X.; Gelovani,J.G.;Qiao,A.;Li,C.Biomacromolecules,2007,8:3422
2 Aliabadi,H.M.;Brocks,D.R.;Lavasanifar,A.Biomaterials, 2005,26:7251
3 Greish,K.;Sawa,T.;Fang,J.;Akaike,T.;Maeda,H.J.Control. Release,2004,97:219
4 Lee,E.S.;Na,K.;Bae,Y.H.J.Control.Release,2005,103:405
5 Attwood,D.;Booth,C.;Yeates,S.G.;Chaibundit,C.;Ricardo,N. M.P.S.Int.J.Pharm.,2007,345:35
6 Yang,Z.L.;Yang,K.W.;Li,X.R.;Liu,Y.Chin.Pharm.J.,2007, 42:519 [杨卓理,杨可伟,李馨儒,刘 艳.中国药学杂志, 2007,42:519]
7 Rijcken,C.J.;Snel,C.J.;Schiffelers,R.M.;van Nostrum,C.F.; Hennink,W.E.Biomaterials,2007,28:5581
8 Calvo,P.;Remuñán-López,C.;Vila-Jato,J.L.;Alonso,M.J. J.Appl.Polym.Sci.,1997,63:125
9 Bravo-Osuna,I.;Ponchel,G.;Vauthier,C.Eur.J.Pharm.Sci., 2007,30:143
10 Peppas,N.A.;Hilt,J.Z.;Khademhosseini,A.;Langer,R.Adv. Mater.,2006,18:1345
11 Xu,Y.;Du,Y.Int.J.Pharm.,2001,250:215
12 Tanima,B.;Susmita,M.;Singh,K.Int.J.Pharm.,2002,243:93
13 Huang,X.L.;Zhang,L.M.J.Function.Polym.,2003,16:594 [黄小龙,张黎明.功能高分子学报,2003,16:594]
14 He,Q.F.;Li,G.M.;Wu,H.Z.;Lu,Z.M.Chin.J.Appl.Chem., 2004,21:192 [何强芳,李国明,巫海珍,卢志敏.应用化学, 2004,21:192]
15 Sorlier,P.;Viton,C.;Domard,A.Biomacromolecules,2002,3: 1336
16 Amiji,M.M.Carbohyd.Polym.,1995,26:211
17 Anthonsen,M.W.;Vårum,K.M.;Hermansson,A.M.;Smidsrod, O.;Brant,D.A.Carbohyd.Polym.,1994,25:13
18 Schatz,C.;Pichot,C.;Delair,T.;Viton,C.;Domard,A.Langmuir, 2003,19:9896
19 Buhler,E.;Rinaudo,M.Macromolecules,2000,33:2098
20 Mladenovska,K.;Cruaud,O.;Richomme,P.;Belamie,E.;Raicki, R.S.;Venier-Julienne,M.C.;Popovski,E.;Benoit,J.P.; Goracinova,K.Int.J.Pharm.,2007,345:59
21 Mi,F.L.;Sug,H.W.;Shyu,S.S.Carbohydr.Polym.,2002,48: 61
22 Li,J.F.;Zhang,L.;Li,J.F.;Zou,Q.;Yang,W.H.;Li,Y.B.Chem. J.Chin.Univ.,2008,29:1874 [李峻峰,张 利,李钧甫,邹 琴,杨维虎,李玉宝.高等学校化学学报,2008,29:1874
23 Ren,D.W.;Wang,Y.L.;Bao,D.C.;Xie,W.Y.;Ma,X.J. Spectros.Spectr.Analy.,2006,26:1825 [任东文,王一力,包德才,谢威扬,马小军.光谱学与光谱分析,2006,26:1825
24 Kim,J.H.;Lee,Y.M.Polymer,1993,34:1952
25 Zeng,R.;Tu,M.;Liu,H.W.;Zhao,J.H.;Zha,Z.G.;Zhou,C.R. Carbohydr.Polym.,2009,78:107
26 Yamada,H.;Teradac,K.;Suryanarayanana,R.J.Pharm. Biomedic.Anal.,2010,51:952
27 Phadnis,N.V.;Cavatur,R.K.;Suryanarayanan,R.J.Pharm. Biomedic.Anal.,1997,15:929
28 Shu,X.Z.;Zhu,K.J.Int.J.Pharm.,2002,233:217
29 Gao,Q.;Wang,G.J.;Li,W.T.Chemistry,2009,72:1 [高 群,王国建,李文涛.化学通报,2009,72:1]
30 Tsaih,M.L.;Chen,R.H.Int.J.Biol.Marcromol.,1997,20:233
31 Shu,X.Z.;Zhu,K.J.;Song,W.H.Int.J.Pharm.,2001,212:19
Preparation and Release Behavior in vitro of Aspirin/Chitosan Nanospheres by Nucleation and Ionic Crosslinking in Emulsion
JIN Shu-Ping FENG Lei*HE Wen-Yan HAN Yu-Qi WEI Yu-Juan
(Key Laboratory of Resources and Environmental Chemistry of West China,Department of Chemistry,Hexi University, Zhangye 734000,Gansu Province,P.R.China)
Chitosan(CS)nanosphere loaded aspirin(aspirin/CS)was prepared by nucleation and ionic crosslinking in an emulsion used for medical and pharmaceutical applications.Chemical component,morphology,size distribution, andcrystalstructureofnanosphereswerecharacterizedbyFouriertransforminfrared(FTIR)spectroscopy,fieldemission scanning electron microscopy(FESEM),transmission electron microscopy(TEM),dynamic laser light scattering (DLLS),and X-ray powder diffraction(XRD).Results showed that the diameter of a typical aspirin/CS nanosphere is about 88 nm and the distribution is uniform.The crystal structure of CS does not change during the nucleation process. The crystallinity of aspirin is dramatically reduced and aspirin is almost amorphous in the nanosphere.The drug content(mass fraction),the drug loading efficiency,and the in vitro release profiles under different conditions were investigated using UV-Vis spectrophotometry.Results showed that the drug content was about 55%,the drug loading efficiency reached 42%,and the chitosan nanosphere displayed an excellent drug-controlled release behavior under the experimental conditions.
Chitosan;Nanosphere;Nucleation in emulsion;Drug-controlled release;Conformation
O645
Received:January 5,2010;Revised:April 26,2010;Published on Web:July 5,2010.
*Corresponding author.Email:flgkl@163.com,zjxjsp@163.com;Tel:+86-936-8282066
ⒸEditorial office of Acta Physico-Chimica Sinica
