高效液相色谱双波长检测法测定维C银翘片中4种组分的含量
2010-10-21孙祥德
董 丽, 孙祥德, 李 琴
(新乡医学院,河南新乡453003)
高效液相色谱双波长检测法测定维C银翘片中4种组分的含量
董 丽*, 孙祥德, 李 琴
(新乡医学院,河南新乡453003)
建立了高效液相色谱测定维C银翘片中维生素C、对乙酰氨基酚、马来酸氯苯那敏和绿原酸4种组分含量的方法。研究优化了样品提取的溶剂与超声提取条件、色谱分离条件和检测波长等,采用了Sinochrom ODS-BP(4.6 mm×200mm,5μm)色谱柱,以0.05mol/L KH2PO4(pH3.0,含1%三乙胺)-乙腈(75∶25,v/v)为流动相,双检测波长260nm和326nm等分离检测条件。结果表明,维生素C、对乙酰氨基酚、马来酸氯苯那敏和绿原酸的测定线性范围均比较宽,方法平均回收率大于99.4%,相对标准偏差(RSD)小于1.8%(n=5),而且测定方法快速、简便、准确,非常适合维C银翘片类药物的质量控制。
高效液相色谱法;双检测波长;维生素C;对乙酰氨基酚;马来酸氯苯那敏;绿原酸;维C银翘片
Abstract:A reversed-phase high performance liquid Chromatographic(RP-HPLC)method with dual wavelength detection was developed for the determination of vitamin C,paracetamol,chlorphenamine maleate and chlorogenic acid in Vitamin C Yinqiao Tablets.The separation was perform ed on a Sinochrom ODS-BP column with the mobile phase consisting of0.05mol/L KH2PO4(pH3.0,containing1%triethylamine and acetonitrile(75∶25,v/v).The detection wavelength was set at260nm(λ1)and326nm(λ2).The method linearities were wide and the average recoveries were more than99.4%.The relative standard deviations were less than 1.8%(n=5).This method is convenient,rap id and accurate.It is readily applied to the quality controls in the production process of Vitamin C Yinqiao Tablets.
Key words:high performance liquid Chromatography(HPLC);dual detection wavelengths;vitamin C;paracetamol;chlorphena mine maleate;chlorogenic acid;Vitamin C Yinqiao Tablets
维C银翘片是由金银花、连翘等13味中药添加维生素C、对乙酰氨基酚和马来酸氯苯那敏3种西药成分所组成的复方制剂,具有辛凉解表、清热解毒之功效,用于治疗流行性感冒引起的发热、头疼、咳嗽、口干、咽喉疼痛等症。目前,国家食品药品监督管理局国家药品标准(WS3-B-4000-98-2003)中仅规定了维生素C和对乙酰氨基酚含量的测定,对其他成分无含量控制标准。金银花是该制剂中中药组方之君药,绿原酸为其主要活性成分,对本方的临床疗效具有重要的作用;马来酸氯苯那敏由于在该制剂中的含量较低,且不易混匀,现行的国家标准也未收载对其含量均匀度的测定。因此,在本制剂中增加绿原酸和马来酸氯苯那敏的含量测定,对全面有效地控制产品质量具有重要的意义。目前,利用光谱法[1-3]、薄层扫描法[4]、毛细管电泳-化学发光法[5]及高效液相色谱法(HPLC)[6-8]等进行维C银翘片中维生素C、马来酸氯苯那敏、对乙酰氨基酚或绿原酸等组分的含量测定多有报道,但这些方法大多是测定其中1种或同时测定其中2种组分。有采用高效毛细管电泳法(HPCE)的胶束电动毛细管色谱(M ECC)模式同时测定对乙酰氨基酚、马来酸氯苯那敏、维生素C和绿原酸4种组分含量的报道[9],但由于HPCE自身的重现性较差,使其难以应用于药品的质量控制。利用HPLC测定维C银翘片中维生素C、对乙酰氨基酚、马来酸氯苯那敏和绿原酸4种组分的含量鲜有报道。本文通过优化样品提取条件和色谱分离条件,用外标法定量,建立了HPLC双波长检测法测定该4种组分含量的方法,并应用于实际样品的测定。方法操作简便,其灵敏度、准确性和重现性均优于已报道的方法。
1 实验部分
1.1 药品与试剂
磷酸二氢钾、磷酸、三乙胺为分析纯试剂,乙腈为色谱纯(天津科密欧化学试剂有限公司)。维生素C、对乙酰氨基酚、马来酸氯苯那敏和绿原酸对照品(中国药品生物制品检定所)。维C银翘片为市售,系3个厂家共9个批次样品(A厂:070386,070345,070320;B厂:070803,070827,070908;C厂:070610,070508,070628;标示量:维生素C49.5 m g/片,对乙酰氨基酚105m g/片,马来酸氯苯那敏1.05m g/片)。实验用水均为自制二次蒸馏水。
1.2 仪器及色谱条件
日本岛津公司的LC-6AD高效液相色谱仪,配LC-solution色谱工作站和SPD-20A紫外检测器等;XK96-A快速混合器(姜堰市新康医疗器械有限公司);pHS-3C精密pH计(上海精密科学仪器有限公司)。色谱柱:大连依利特公司Sinochrom ODS-B P(200mm×4.6mm,5μm),预柱:C18(25 mm×4.6mm);流动相:0.05mol/L KH2PO4(pH 3.0,含1%三乙胺)-乙腈(75∶25,v/v);检测波长:260nm和326nm;流速:1.0mL/m in;柱温:25℃;进样量:20μL;用外标法定量。
1.3 样品溶液的制备
取药品20片,除去包衣,精密称定,计算平均片重。粉碎研磨后精密称取适量(约相当于1片的质量),置于100mL容量瓶中,加流动相适量,间歇超声处理使之溶解,用流动相稀释至刻度,摇匀,过滤,续滤液作为供试品溶液A,用于测定马来酸氯苯那敏和绿原酸。取供试品溶液A2.50mL置于100 mL容量瓶中,用流动相稀释至刻度,得供试品溶液B,用于测定维生素C和对乙酰氨基酚。
1.4 对照品溶液的制备
分别准确称取维生素C、对乙酰氨基酚、马来酸氯苯那敏和绿原酸对照品适量,以流动相溶解,配制4组分对照品储备液,其质量浓度分别为1.030,1.020,1.090和0.500 0g/L。量取维生素C、对乙酰氨基酚、绿原酸3种储备液各5.00mL,马来酸氯苯那敏储备液10.00mL置于同一50mL容量瓶中,用流动相稀释至刻度,得4组分混合对照品溶液,混合溶液中各组分的质量浓度分别为维生素C 103.0m g/L,绿原酸50.00m g/L,对乙酰氨基酚102.0m g/L,马来酸氯苯那敏218.0m g/L。将此混合对照品溶液用流动相依次等体积稀释,得到系列质量浓度的混合对照品溶液。
2 结果与讨论
2.1 样品溶液制备方法的选择
马来酸氯苯那敏和维生素C在水中易溶,而绿原酸和对乙酰氨基酚在水中溶解度较小。根据4组分的溶解性,本文曾试验以水、甲醇、不同比例的甲醇-水及本文最终选定的流动相等不同的提取溶剂进行提取。结果显示,用水提取时,对乙酰氨基酚和绿原酸提取不完全,而合适比例的甲醇-水混合溶剂虽能对各被测组分有较好的提取率,但提取的共存成分较多,干扰被测组分的分离。用流动相提取不仅提取完全,而且干扰组分少,又不产生溶剂峰。因此,本文选择用流动相作提取溶剂。同时还试验了室温下浸取、振荡、超声提取等不同的提取方式。结果表明,超声提取简便快速、提取完全;但由于维生素C对热不稳定,超声所产生的热量会使其破坏,从而影响测定结果的准确度。同时,绿原酸在酸性介质(pH2~4)中的稳定性较好[10],但在较高温度下会加速其分子中酯键的水解。因此,为确保提取过程中维生素C和绿原酸不被破坏且提取完全,本文采用间歇超声提取的方式。
2.2 检测波长的选择
以流动相为空白,在200~400nm范围内扫描,维生素C、对乙酰氨基酚、绿原酸和马来酸氯苯那敏的最大吸收波长分别为245,249,326和260 nm。由于绿原酸与其他3组分的最大吸收波长相差悬殊,故采用双波长同时检测模式进行检测。该制剂中马来酸氯苯那敏的处方量甚小,故选择260 nm作为测定的第一检测波长,在此波长下,维生素C和对乙酰氨基酚也具有较大的吸收,检测灵敏度足以达到分析要求。以绿原酸的最大吸收波长326 nm作为第二检测波长对绿原酸进行检测。双波长检测模式兼顾了各组分的含量差别和检测灵敏度,实现了在同一色谱条件下4组分含量的同时测定。
文献[11]在以HPLC测定维C银翘片中的上述4组分的含量时,选择219nm检测维生素C、对乙酰氨基酚和马来酸氯苯那敏,尽管3组分在219 nm波长下具有较强吸收,但由于维C银翘片中含有大量的中药成分,基质复杂,经溶剂提取制备的供试品溶液中含有大量的有末端吸收的共存成分,从而干扰被测组分的分离测定。另外,该文献使用甲醇-水-冰醋酸(体积比为20∶80∶0.2)为提取溶剂,冰醋酸在219nm处也有吸收,从而产生溶剂峰。本文结果显示,选择在260nm处检测维生素C、对乙酰氨基酚和马来酸氯苯那敏3组分,不仅检测灵敏度足以达到分析要求,而且有效地减少了大量具有末端吸收的未知共存成分的干扰。
2.3 流动相的选择
马来酸氯苯那敏是氯苯那敏的马来酸盐,将出现马来酸和氯苯那敏两个色谱峰。本实验表明,流动相中乙腈的比例对绿原酸和氯苯那敏的保留行为影响很大,调整乙腈和缓冲盐的比例,可实现各组分的分离。流动相中加入少量的三乙胺不仅可以调节各组分的保留时间,而且可很好地改善氯苯那敏的峰形。曾试验不同pH值的缓冲溶液对各待测组分分离的影响,结果显示,改变流动相的pH值对维生素C、马来酸、绿原酸和对乙酰氨基酚的保留时间几乎没有影响,而氯苯那敏的保留时间则随着pH值的降低而缩短。这是由于氯苯那敏分子中含有叔氨基,溶液的酸度增加有利于该氨基的质子化,从而使其极性增大,导致其在反相色谱柱上的保留减弱。尽管酸度的降低有利于氯苯那敏较早出峰,但ODS柱难以承受pH2以下的酸度,且维生素C在过高的酸度下不稳定;而绿原酸分子结构中含有的酯键,其水溶液在中性和碱性环境下稳定性较差,但在pH 2~4下较稳定。经优化,最终确定流动相为0.05 mol/L KH2PO4(pH3.0,含1%三乙胺)-乙腈(75∶25,v/v)。在本文选定的色谱条件下,经用不含待测组分的空白样品进行试验,试样中提取的未知组分不干扰待测组分的测定。
目前,采用HPLC分离分析马来酸氯苯那敏的报道很多,但有不少文献报道的测定条件[12-16]值得商榷。由于马来酸的极性较大,而氯苯那敏的极性较弱,所以马来酸在反相色谱上的保留弱于氯苯那敏。流动相中有机溶剂相的比例对氯苯那敏的保留时间影响甚大,当有机相的比例较低时,氯苯那敏常常未被洗脱。因此,有不少文献常将较早洗脱的马来酸峰误认为是氯苯那敏峰而进行定量[16]。由于马来酸氯苯那敏的药效结构是氯苯那敏,因此在进行马来酸氯苯那敏的含量测定中以马来酸峰来定量是不妥的。文献[11]采用了以甲醇-乙腈-0.02%磷酸(5∶10∶85,v/v)为流动相,柱温为35℃,检测波长为310nm和219nm的色谱条件。但经验证发现在该文献的色谱条件下,氯苯那敏在35m in以后出峰(这是由于流动相中甲醇、乙腈比例偏低所致),而用于定量马来酸氯苯那敏的峰系马来酸峰,且以末端吸收219nm为检测波长,干扰成分较多。本实验改进并优化了流动相条件和检测波长,使维生素C、绿原酸、对乙酰氨基酚和氯苯那敏4组分在10m in内全部出峰并得到了很好的分离。方法学验证结果显示,本文方法测定维C银翘片4组分快速、简便、准确。
在本文确定的色谱条件下,4组分之间以及与其他未知组分之间得到了很好的分离,以对乙酰氨基酚计,柱效不低于3 000。混合对照品溶液和样品溶液的色谱图分别见图1和图2。
2.4 线性范围与定量限确定
分别取系列对照品混合液20μL进样,按照1.2节色谱条件测定,以各组分峰面积A对相应的质量浓度C回归制备标准曲线。当信噪比(S/N)为10时,确定4种化合物的定量限。结果(见表1)表明,4种组分的峰面积与进样浓度之间线性关系良好,线性范围宽,灵敏度高。

图1 双检测波长下4组分混合对照品溶液的色谱图Fig.1 HPLC chromatograms of the standard solution at the dual detection wave lengths

图2 双检测波长下供试品溶液的色谱图Fig.2 HPLC chromatogram s of the sample solutions at the dual detection wave lengths

表1 4种组分的回归方程、相关系数、线性范围与定量限(S/N=10)Table1 Regression equations,correlation coefficients,linear ranges and limits of quantification(LOQ,S/N=10)of four components
2.5 精密度试验
配制4种组分高、中、低3个浓度的对照品溶液,其中维生素C溶液的质量浓度分别为0.201 1,6.438,103.0m g/L;对乙酰氨基酚溶液的质量浓度分别为0.199 2,3.188,51.00m g/L;绿原酸溶液的质量浓度分别为0.097 60,3.125,50.00m g/L;马来酸氯苯那敏溶液的质量浓度分别为0.425 5,13.62,218.0m g/L。上述各溶液重复进样5次进行含量测定及日内精密度(以相对标准偏差(RSD)计)测定;每种溶液连续5d配制测定,计算日间精密度。结果表明,各组分高中低3种浓度溶液的日内精密度均小于1.0%,日间精密度均小于1.8%。
2.6 回收率试验
称取约1片质量的已知含量的样品(A厂,070386批号)粉末5份,各准确加入含有维生素C 10.00m g、对乙酰氨基酚20.00m g、绿原酸1.00 m g和马来酸氯苯那敏1.00m g的4组分对照品储备液适量,置于100mL容量瓶中,用流动相超声助溶,定容至刻度。过滤,取续滤液进样20μL测定马来酸氯苯那敏和绿原酸。取续滤液2.50mL于100 mL容量瓶中,用流动相稀释至刻度,进样20μL测定维生素C和对乙酰氨基酚,测定结果见表2。

表2 4种组分的回收率测定结果(n=5)Table2 Recovery results of four components(n=5)
2.7 溶液的稳定性试验
取新配制的样品供试溶液于室温下和4℃冰箱中放置,并分别于放置0,1,3,5,7,12,24h取样20μL进行测定。结果表明,室温下对乙酰氨基酚、绿原酸和马来酸氯苯那敏峰面积的RSD分别为0.94%,2.54%和2.67%,表明这3种组分在24h内有较好的稳定性;但室温下维生素C放置3h峰面积即降低了约10%。而放置在4℃冰箱内保存,24h内4组分的峰面积的RSD均小于2.0%。
2.8 耐用性试验
根据《中国药典》2005版药品质量标准分析方法验证指导原则,考察了方法的耐用性。按照已确定的色谱条件进行试验,同一样品使用同一色谱柱(Spherigel C18柱,200mm×4.6mm,10μm)在3台不同仪器(Sh im adzu LC-6A,Shim adzu LC-6AD,Varian PROSTAR210)上测定;使用同一台仪器(Sh im adzu LC-6AD),分别用Sinochrom ODS-B P(200mm×4.6mm,5μm)、Shim adzu VP-ODS(250mm×4.6mm,5μm)和Spherigel C18(200 mm×4.6mm,10μm)3根不同厂家的色谱柱,在不同时间经不同实验人员测定。结果显示,4组分的峰形均较理想,相邻峰的分离度均大于1.5,理论塔板数均大于3 000,测定结果的RSD小于2.6%。另外还考察了柱温和缓冲液pH值等因素的微小变化对分离度和峰面积的影响。结果显示,当柱温在±5℃范围内波动时,测定结果无明显的差异。分别用pH值为2.80,2.90,3.10的缓冲液配制流动相进行试验,其实验结果和分离度无明显的差异。上述试验结果显示,本方法在测定条件有较小的变动时,方法的耐用性良好。
2.9 样品测定
取3个厂家的9批次维C银翘片样品按1.3节下的方法操作,取供试品溶液20μL进样分析,其中马来酸氯苯那敏以氯苯那敏峰的峰面积进行定量,结果见表3。
由于该制剂处方中马来酸氯苯那敏和绿原酸的含量相对较低,用同一色谱图定量会存在着部分组分的浓度不在其线性范围内,故在样品测定时先制成供试品溶液A供测定马来酸氯苯那敏和绿原酸,然后将供试品溶液A适当稀释供测定维生素C和对乙酰氨基酚。
表3的测定结果显示,不同生产厂家不同批次的维C银翘片中的对乙酰氨基酚的含量符合规定,少数产品维生素C含量不均匀,而马来酸氯苯那敏和绿原酸两成分的含量差异较大。因此,规范稳定药材来源,在该制剂质量标准中增加马来酸氯苯那敏和绿原酸含量的测定项目,对保证维C银翘片的质量、安全、有效具有重要的意义。
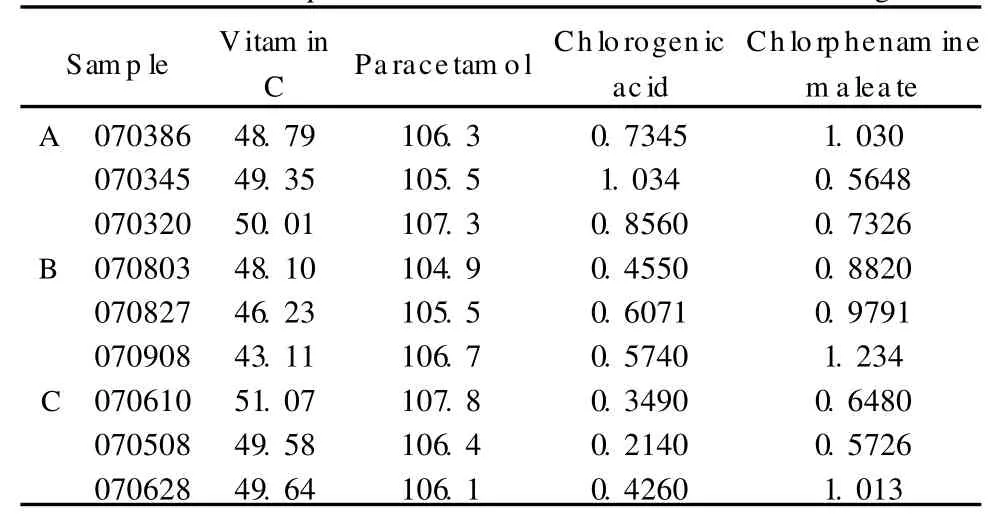
表3 不同批次维C银翘片中4组分的含量(n=3)Table3 Contents of four components in vitamin C Yinqiao Table ts(n=3)m g/tablet
3 结论
本文应用HPLC-双波长检测建立了同时高灵敏度地测定维C银翘片中4种组分的含量方法,并进行了方法学验证。在测定含有马来酸氯苯那敏的复方制剂的多组分含量时,不仅需要优化色谱条件使各待测组分不受干扰,还应特别注意避免将马来酸峰误认为氯苯那敏进行定量。本文方法简便、快速、灵敏度高、重现性好,可用于维C银翘片的质量控制。
[1] Lu W,Zhao X J.Chinese Pharmaceutical Affairs(陆伟,赵新静.中国药事),2004,18(7):435
[2] Cai Z,Zhao J,Du L W,et al.Applied Chemical Industry(蔡卓,赵静,杜良伟,等.应用化工),2008,37(10):1235
[3] Bai H J,Zhang L,Huang Z S.Tianjin Pharm acy(白海娇,张莉,黄哲甦.天津药学),2007,19(3):10
[4] J iang J,Zhao H Q,Yang L,et al.Journal of Shenyang Pharm aceuticalUniversity(姜静,赵怀清,杨丽,等.沈阳药科大学学报),2002,19(6):417
[5] W ang J W,Peng Z B,Yang J.Chinese Journal of Analysis Laboratory(汪敬武,彭志兵,杨佳.分析试验室),2007,26(4):25
[6] Xue P,Lu J,Cai J,et al.D rug Standards of China(薛萍,陆骏,蔡健,等.中国药品标准),2006,7(1):27
[7] Fan Q M,Sun D Y,Zhou H J.Research and Practice of Chinese Medicines(范全民,孙冬云,周慧娟.现代中药研究与实践),2005,19(6):43
[8] Guo F L,Zhao X J.Chinese Journal of Pharmaceutical Analysis(郭凡岭,赵新静.药物分析杂志),2005,25(2):223
[9] Yin C,Wu Y T.Chinese Journal of Pharmaceutical Analysis(尹茶,吴玉田.药物分析杂志),2000,20(1):24
[10] Lin L Y,He Y J,Luo W W,et al.West China Journal of Pharmaceutical Sciences(林丽洋,贺英菊,罗巍伟,等.华西药学杂志),2005,20(3):225
[11] Gao G W,Feng X D,Huang H X.Chinese Traditional Patent Medicine(高光伟,冯向东,黄海欣.中成药),2008,30(2):207
[12] W an L,Wu R.China Pharmacist(万莉,伍蓉.中国药师),2009,12(5):671
[13] Lou Z H.D rug Standards of China(娄志红.中国药品标准),2007,8(6):65
[14] J i Y.D rug Standards of China(纪宇.中国药品标准),2004,5(1):31
[15] Xu Y,Liu F,Xu X X,et al.Sichuan Journal of Physiological Sciences(徐燕,刘峰,徐晓霞,等.四川生理科学杂志),2008,30(3):110
[16] Sun S F,Liu G R,Wang Z,et al.Chinese Journal of Pharmaceuticals(孙素芳,刘国瑞,王喆,等.中国医药工业杂志),2006,37(9):627
Determination of four components in Vitamin C Yinq iao Tablets using reversed-phase high performance liquid chromatography with dual wave length detection
DONG L i*,SUN Xiangde,LI Q in
(Xinxiang Medica l University,Xinxiang 453003,China)
O658
A
1000-8713(2010)02-0204-05
*通讯联系人:董 丽,副教授,主要研究方向为药物色谱分析.Tel:(0373)3029128,E-m ail:ldong@xxmu.edu.cn.
新乡医学院高学历人才基金项目(No.2005YJ31).
2009-07-20
DO I:10.3724/SP.J.1123.2010.00204
