动物性食品中硝基呋喃类药物残留检测研究进展
2010-09-26祝伟霞刘亚风
祝伟霞,刘亚风,梁 炜
(河南出入境检验检疫局,河南郑州 450003)
硝基呋喃类药物是人工合成的具有5-硝基基本结构的广谱抗菌药物[1],是一类具有潜在致癌和诱导有机体产生突变的物质。硝基呋喃类药物主要包括呋喃唑酮、呋喃西林、呋喃妥因、呋喃它酮(图1)[2-3]。硝基呋喃类药物常用于治疗和预防沙门菌、大肠埃希菌感染引起的猪、鱼、禽类消化系统感染。该类药物半衰期很短,在动物体内能迅速代谢[4],与蛋白结合的代谢物能产生稳定的残留,常用的食品烹饪方法如蒸煮、烘烤和微波加热等均无法使代谢物降解[5]。Chu P S等[6]研究鲶鱼口服呋喃类药物时发现,其代谢物在56 d后检测还呈阳性。采用同位素标记对呋喃唑酮的毒性进行研究,证明该药具有致癌和致基因突变性,蛋白结合态的呋喃唑酮代谢物含有3-氨基-2-唑酮的完整侧链,在胃液的弱酸条件下,侧链可以从结合态的母体分子中解离,水解为具有致癌性的基团β-羟乙基肼[7]。
欧盟2377/90/EEC已全面禁止呋喃类抗菌药物作为生长剂和杀菌剂用在动物饲料中,欧盟要求以代谢物为目标分析物,检测硝基呋喃原药残留,并要求方法灵敏度要达到1μg/kg[8],同时欧盟1756/2002/EC也禁用了新发现的呋喃类药物硝呋索尔。2002年我国出口到欧盟的虾查出硝基呋喃阳性,欧盟通报所有出口国检测呋喃类药物[9],中国、美国、日本也相继禁止在食用动物中使用呋喃类药物作为生长剂和杀菌剂,方法检出限要求达到0.5μg/kg[10]。为了应对“绿色壁垒”的挑战,本文全面综述了目前硝基呋喃类代谢物残留检测的样品前处理方法、仪器测定方法及呋喃西林假阳性的问题。
1 样品前处理
1.1 样品水解和衍生化
硝基呋喃类抗生素在动物体内代谢快,半衰期短。血浆、尿不能用于呋喃类代谢物检测,一般选用禽肉、鸡蛋、肝脏等动物性食品为检测对象,原药代谢后以蛋白结合物形态存在于机体组织中,根据试验目的可分为硝基呋喃代谢物总量和结合态代谢物检测。Conneely A等[11]测定结合态呋喃唑酮代谢物时样品先用甲醇、水、乙醇、乙醚洗涤,以除去游离态代谢物。结合态代谢物只有在适当的酸性条件下才能释放出来,硝基呋喃检测中常用稀盐酸溶液解离结合态代谢物。4种硝基呋喃代谢物分子质量均在75 g/mol~201 g/mol[12],进行液相色谱串联质谱测定时必须进行衍生化,衍生化是为了增加分子质量,使化合物不在高噪音背景下,增加特征碎片离子的选择性[3],提高质谱响应。常采用2-硝基苯甲醛(2-NBA)衍生化氨基增加代谢物的离子化效率,水解后生成硝基苯衍生物,衍生化一般在37℃过夜进行,亲核基团R-NH2酸性催化环境很快游离出来与2-NBA碳酰基发生化学作用。
M ottier P等[8]在鸡肉中添加5μg/kg硝基呋喃代谢物进行衍生化时发现 1-氨基-2-内酰腺(AHD)和氨基脲(SEM)最稳定,而 3-氨基-2-唑烷基酮(AOZ)和 5-甲基吗啉-3-氨基-2-唑烷基酮(AMOZ)损失40%。Leitner A等[13]等采用不同衍生化试剂对代谢物衍生化效率及稳定性进行比较,结果表明,2-NBA是呋喃类代谢物最有效的衍生化试剂,衍生化效率大于70%。李耀平等[14]建立在亲核加成反应机理的基础上,采用新衍生剂2-氯苯甲醛衍生硝基呋喃代谢物,提高了灵敏度,缩短了衍生反应时间。
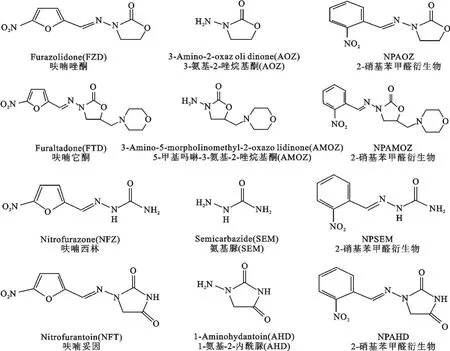
图1 4种硝基呋喃原药、代谢物、衍生物结构式Fig.1 The stuctu res of four nitrofu rans inc luding medicines,m atabolites,derivates
1.2 液液萃取净化
呋喃类液液萃取一般是在pH 6~7水溶液与乙酸乙酯进行两相分配[15-16],在液液萃取操作过程中,样品溶液的pH对萃取效率有很大的影响,丁涛等[17]试验不同pH对回收率的影响,发现pH为7~7.5时,AHD、AOZ、SEM 和AMOZ代谢物的衍生产物提取效率最高,分别为92.1%、95.1%、91.4%和94.3%。液液萃取易产生乳化现象,特别对含高蛋白、高脂肪、高淀粉的样品,需要加入冗长的去乳步骤,延长了试验操作时间。
1.3 固相萃取净化
为了更有效地净化动物源性食品中硝基呋喃类代谢物,常采用阴离子交换混合机理柱(MAX)、聚苯乙烯-二乙烯苯柱(LiChrolut EN)和亲水-亲脂平衡料固相萃取柱(HLB)净化样品[1-2,18],利用pH 7时分子间π-π作用力保留硝基芳香化合物,有机溶剂如乙酸乙酯或甲醇破坏这种作用力而洗脱被测物,Conneely A等[11]分别试验了溶液pH为3、6和10时H LB小柱净化时的回收率,数据表明pH 6时HLB柱回收率最高。林黎明等[1]评价了 MAX与HLB串联柱、LiChrolut EN柱、HLB柱净化时的回收率,最后得出H LB柱与EN柱获得相同的回收率。
2 检测方法
呋喃类结构化合物紫外吸收不明显,因此在紫外检测时灵敏度很低,不能满足代谢物0.5μg/kg限量要求;同时毛细管电泳和薄层色谱因其灵敏度低,在呋喃类代谢物检测中应用很少[3,19],其代谢物残留检测常用液相色谱串联质谱法和酶联免疫法。
2.1 液质联用
液质联用技术同时利用了液相色谱较强的分离能力与质谱检测器丰富的结构信息,为目标化合物提供了可靠的定性和定量结果。由于硝基呋喃类药物代谢物具有较低的离子化效率并缺少特征离子,2-NBA衍生物增加了相对分子质量,提高了离子化效率。欧盟EEC/657/2002指令规定对于禁用药物的质谱确证方法必须达到4个确证点。在串联质谱分析时,检测1个母离子和2个子离子能达到4个确证点的要求。液质联用测定呋喃类代谢物液相分离时流动相中常用有机溶剂(甲醇,乙腈)与弱酸(甲酸,乙酸,甲酸铵,乙酸铵)的混合液。正离子时加入酸能促使目标物的离子化,增强检测灵敏度。目前液相色谱-串联质谱联用已用于测定动物组织[1,8,11,13]、牛奶[12]、禽组织[10,16]、鸡蛋[5,15]、鲶鱼[6]、蜂王浆[17]、蜂蜜[2]等动物性食品基质中硝基呋喃类代谢物残留量,检出限在0.1μg/kg~2μg/kg[20]。
2.2 酶联免疫
酶联免疫法(ELISA)是一种特异性强、灵敏度高的技术,常作为筛选方法用于残留分析中。该方法能快速、简单地测定1种硝基呋喃代谢物残留,灵敏度取决于制备抗体的选择性。Vass M等[21]采用ELISA快速测定鸡蛋中呋喃西林代谢物,样品经酸衍生化后酶联免疫反应,方法EEC/657/2002验证,检出限(CCα)0.13 μg/kg,定量限(CCβ)0.3 μg/kg,回收率在77.8%~110%,阳性测定结果与液质比较,表明该方法测定结果准确;Chang C等[22]采用ELISA测定可食性动物组织呋喃唑酮代谢物时以苯甲醛作为衍生化试剂,生成苯AOZ的衍生物进行检测,回收率55.8%~99.6%。Pim pitak U等[23]采用ELISA测定虾中AMOZ,检出限为0.16μg/kg。
3 SEM假阳性问题
近年来,SEM阳性检出率越来越高,在被检出的呋喃类阳性结果中47%是SEM阳性,研究发现,SEM的污染来自三种不同渠道[24],即呋喃类兽药残留、卡拉胶和偶氮二甲酰胺。卡拉胶常用作食品稳定剂和稠化剂,是红海藻中萃取出的一种多聚糖;偶氮二甲酰胺常作为发泡剂和面粉添加剂用于食品中,SEM也是偶氮二甲酰胺的一种降解产物,2005年欧盟已禁止其作为发泡剂用于食品。
4 硝基呋喃类药物检测存在问题及展望
目前动物性食品中硝基呋喃类药物检测存在着样品前处理繁琐,试剂用量大,检测仪器昂贵,SEM
阳性率高等问题。呋喃类检测的研究重点是在不需要衍生化步骤的情况下,直接检测其代谢物来减少分析时间和成本。呋喃类药物残留研究的另一方向是建立快速ELISA法,但目前商品化试剂盒只有单克隆抗体,若成功开发出4种呋喃类药物的抗体检测试剂盒,则能使硝基呋喃代谢物检测达到简便、快速、成本低廉、特异性强的目的。
[1]林黎明,林回春,刘心同,等.固相萃取高效液相色谱-质谱法测定动物组织中硝基呋喃代谢产物[J].分析化学,2005,33(5):707-710.
[2]Lopez M I,Feldlaufer M F,W illiams A D,et al.Determination and confirmation of nitrofuran residues in honey using LC-MS/MS[J].JAgric Food Chem,2007,55:1103-1108.
[3]Wickramanayake P U,T ran T C,H ughes JG.Simultaneous separation of nitrofu ran antibiotics and their metabolites by using m icellar electrokinetic capillary chromatography[J].E-lectrophoresis,2006,27:4069-4077.
[4]蒋 原,丁 涛,徐锦忠.硝基呋喃类药物在克氏螯虾组织中消除规律的研究[J].畜牧与兽医,2008,40(2):34-37.
[5]Finzi JK,Donato JL,Sucupira M,et al.Determination of nitrofuran metabolites in poultry musc le and eggs by liquid ch romatography-tandem mass spectrom etry[J].JChromatog r B,2005,824:30-35.
[6]Chu P S,Lopez M I,Abraham A,et al.Residue depletion of nitrofuran drugs and their thissue-boundmetabolites in channel catfish after oral dosing[J].J Agric Food Chem,2008,56:8030-8034.
[7]Barbosa J,Ferreira mL,Ramos F,et al.Determination of the furaltadone metabolite 5-methy lmorpholino-3-amino-2-oxazolidinone(AMOZ)using liquid ch romatography coupled to electrosp ray tandem m ass spectrom etry during the nitrofuran crisis in Portugal[J].Accred Qual Assur,2007,12:543-551.
[8]M ottier P,Khong SP,Gremaud E,et al.Quantitative determination of fou r nitrofuranm etabolites in meat by isotope dilution liquid chromatography-electrospray ionisation-tandem mass spectrometry[J].JChromatog r A,2005,1067:85-91.
[9]H oenicke K,Gatermann R.H ow can zero toleran ces be controlled the case study of nitrofu rans[J].Accred Qual Assu r,2006,11:29-32.
[10]Verdon E,Couedor P,Sanders P.M ulti-residuem onitoring for the simultaneous determination of five nitrofu rans(fu razolidone,furaltadone,nitrofurazone,nitrofurantoine,nifu rsol)in poultry muscle tissue through the detection of their fivemajor metabolites(AOZ,AMOZ,SEM,AHD,DNSAH)by liquid chromatog raphy coupled to electrospray tandem mass spectrom etry-in-house validation in line w ith Comm ission Decision 657/2002/EC[J].Anal Chim Acta,2007,586:336-347.
[11]Conneely A,Nugent A,Keeffe M,et al.Isolation of bound residues of nitrofu ran drugs from tissue by solid-phase extraction w ith determination by liquid ch romatography w ith UV and tandem mass spectrom etric detection[J].Anal Chim Acta 2003,483:91-98.
[12]Rodziewicz L.Determination of nitrofu ran metabolites in m ilk by liquid ch romatography-electrosp ray ionization tandem mass spectrom etry[J].JChromatogr B,2008,864:156-160.
[13]Leitner A,Zollner P,Lindner W.Determination of the metabolites of nitrofuran antibiotics in animal tissue by high-performance liquid ch rom atography-tandem mass spectrometry[J].JCh rom atogr A,2001,939:49-58.
[14]李耀平,林永辉,贾东芬,等.水产品中硝基呋喃代谢物残留快速检测新方法的研究[J].分析测试学报,2008,27(7):712-717.
[15]Bock C,Stachel C,Gowik P.Validation of a confirmatory m ethod for the determination of residuesof fou r nitrofurans in egg by liquid chromatography-tandem mass spectrometry w ith the softw are Inter Val[J].Anal Chim Acta,2007,586:348-358.
[16]Bock C,Gowik P,Stachel C.M atrix-comprehensive in-house validation and robustness check of a confirmatory method for the determination of fou r nitrofuran metabolites in pou ltry m uscle and shrim p by LC-MS/MS[J].J Ch rom atogr B,2007,856:178-189.
[17]丁 涛,徐锦忠,沈崇钰,等.高效液相色谱-串联质谱联用测定蜂王浆中的四种硝基呋喃类药物的代谢物[J].色谱,2006,24(5):432-435.
[18]Xia X,LiXW,Zhang SX,etal.Simultaneousdetermination of 5-nitroim idazoles and nitrofu rans in pork by high-performance liquid chromatog raphy-tandem m ass spectrometry[J].J Chromatogr A,2008,1208:101-108.
[19]Berezkin V G,Onu chak L A,Evtyugina E N.Capillary thinlayer chroam tog raphy of antibacterial nitrofu ran derivates[J].Russian JAppli Chem,2009,82(2):312-316.
[20]祝伟霞,杨冀州,魏 蔚,等.高效液相色谱串联质谱法测定动物源性食品中硝基呋喃类代谢物残留[J].现代畜牧兽医,2008(1):47-50.
[21]Vass M,Dib likova I,Kok E,et al.In-house validation of an ELISA method for screening of sem icarbazide in eggs[J].Food Addi Conta,2008,25(8):930-936.
[22]Chang C,Peng D P,Wu JE,et al.Development of an indirect competitive ELISA for the detection of fu razolidone marker residue in anim aledible tissues[J].JAg ric Food Chem,2007,56:1525-1531.
[23]Pim pitak U,Putong S,Komolpis K.Developmen t of am onoc lonal antibody-based enzym e-linked immunosorben t assay for detection of the fu raltadone metabolite,AMOZ,in fortified shrimp samples[J].Food Chem,2009,116:785-791.
[24]Szilagyi S,Calle B.Developm ent and validation of an analyticalmethod for the determination of sem icarbazide in fresh egg and in egg powder based on the use of liquid chromatography tandem m ass spectrometry[J].Anal Chim Acta,2006,572:113-120.
